Ardelyx
Company overview
Ardelyx (ARDX) is a specialized biopharmaceutical company focused on developing first-in-class, disruptive medicines for the treatment of renal diseases. The company's primary therapeutic focus is on treating people with renal diseases, which affect both the heart and the kidneys. This includes patients with end-stage renal disease, or ESRD, who suffer from elevated serum phosphorus, or hyperphosphatemia; patients with chronic kidney disease, or CKD, and/or heart failure patients with elevated serum potassium, or hyperkalemia. Ardelyx has also developed a number of programs directed toward treating gastrointestinal, or GI, disorders, including the treatment of irritable bowel syndrome with constipation, or IBS-C.
The company's portfolio is led by the development of tenapanor, a first-in-class inhibitor of NHE3. In its renal pipeline, tenapanor is being evaluated in a second Phase 3 trial for the treatment of hyperphosphatemia in patients with ESRD who are on dialysis. This registration trial follows a successful first Phase 3 trial completed in 2017, which achieved statistical significance for the primary endpoint. Ardelyx is also advancing a small molecule potassium secretagogue program, RDX013, for the potential treatment of hyperkalemia. The company believe that both tenapanor and RDX013 have the potential to provide treatment options that are differentiated significantly from binders, the current standards of care in both of these markets. The company believe its small molecule approach to treating these conditions could significantly reduce the pill burden for patients, leading to higher compliance, and offer completely new mechanisms of action that interact with receptors in the gut, potentially allowing improved efficacy in some patients.
In addition to the development for renal diseases, Ardelyx has developed tenapanor for the treatment of patients with IBS-C. In 2017, the company completed the T3MPO program for this indication, including two Phase 3 studies, both of which achieved statistical significance for the primary endpoint, and a long-term safety extension study. The company believe that data from the T3MPO program collectively demonstrated the ability of tenapanor to provide sustained relief of constipation and reduced abdominal pain with a generally favorable tolerability profile. Based on the results of the T3MPO clinical program in IBS-C, Ardelyx is preparing to submit its first New Drug Application, or NDA, to the United States Food and Drug Administration, or FDA, in the second half of 2018 for tenapanor for the treatment of IBS-C.
Commercial Strategy
The company aim to build a multi-product company that commercializes its renal products in the United States. The company's strategy is to leverage ex-U.S. collaborations with established industry leaders to efficiently bring its renal medicines to patients outside the United States. Additionally, its goal is to bring tenapanor for IBS-C to market by leveraging domestic and ex-U.S. collaborations.
In November 2017, the company entered into a license agreement that provides Kyowa Hakko Kirin Co., Ltd., or KHK, with exclusive rights to develop and commercialize tenapanor for the treatment of cardiorenal diseases, including hyperphosphatemia, in Japan. Under the terms of the license agreement, the company received a $30 million upfront payment and are eligible to receive up to $130 million in development and commercialization milestones based upon currency exchange rates as of the effective date of the license agreement, as well as high-teen royalties on net sales throughout the term of the agreement.
In December 2017, the company entered into a license agreement with Shanghai Fosun Pharmaceutical Industrial Development Co. Ltd., or Fosun Pharma, providing Fosun Pharma with the exclusive rights to develop and commercialize tenapanor in China for the treatment of patients with hyperphosphatemia related to CKD and patients with IBS-C. Under the terms of the agreement, the company received an upfront payment of $12 million and are eligible to receive additional milestones of up to $113 million, as well as tiered royalties on net sales ranging from the mid-teens to 20%.
Proprietary drug discovery and design platform
In line with its overall strategy and transition to focus solely on its renal pipeline, Ardelyx has shifted its research focus to support its preclinical and clinical development candidates including tenapanor and RDX013, as well as other potential renal opportunities. The company intend to continue to utilize its unique discovery and design platform to help elucidate first-in-class mechanisms of action, as with tenapanor, and to inform preclinical experiments to help advance its product candidates. The company also use its platform to further support the potential commercialization of its programs, which is valuable to it and future partners. The company believe that its platform, and early pipeline, represent additional collaborative opportunities, market potential and downstream value-creation.
Using its platform, Ardelyx has been able to discover targets found in the GI tract that regulate important processes in the body and design products candidates that act upon those targets to take advantage of the gut’s ability to communicate with other organs. The company's platform integrates two critical concepts: ![]() its proprietary chemistry capabilities that enable it to design and optimize gut-restricted compounds that can provide a higher margin of safety than systemically absorbed compounds, and (ii) its stem cell-based translational technology called the Ardelyx Primary Enterocyte and Colonocyte Culture System, or APECCS, that enables it to discover targets in the GI tract which control health and disease processes, to optimize drug candidates and to understand their mechanisms of action. The company's platform can be applied across the entire GI tract, allowing for the broadest evaluation of disease targets to develop medicines optimized for specific diseases. The predictive ability of its platform enables it to better assess, at a very early stage, the potential for small molecule compounds to treat specific diseases.
its proprietary chemistry capabilities that enable it to design and optimize gut-restricted compounds that can provide a higher margin of safety than systemically absorbed compounds, and (ii) its stem cell-based translational technology called the Ardelyx Primary Enterocyte and Colonocyte Culture System, or APECCS, that enables it to discover targets in the GI tract which control health and disease processes, to optimize drug candidates and to understand their mechanisms of action. The company's platform can be applied across the entire GI tract, allowing for the broadest evaluation of disease targets to develop medicines optimized for specific diseases. The predictive ability of its platform enables it to better assess, at a very early stage, the potential for small molecule compounds to treat specific diseases.
Product Pipeline
Tenapanor: A New Approach for Treating Hyperphosphatemia in ESRD Patients on Dialysis
The lead product candidate in its renal portfolio is tenapanor for the treatment of hyperphosphatemia, or high levels of blood phosphorus, in ESRD patients on dialysis. Hyperphosphatemia is a significant problem among dialysis patients worldwide.
CKD is the progressive deterioration of renal function that can occur over several months or years. The symptoms of worsening kidney function are nonspecific, and can include having less energy, reduced appetite, dry itchy skin, swollen feet and ankles or generally just not feeling well. If the deterioration continues and is not halted by changes in lifestyle or with the assistance of pharmacological intervention, the disease will likely cause significant cardiovascular morbidity, and can progress to ESRD, the final stage of CKD, where kidney function will be lost entirely.
Current management of ESRD includes hemodialysis and peritoneal dialysis as a means to filter toxins from the blood once kidneys have failed. Unless this intervention occurs, kidney failure results in the accumulation of waste products that may ultimately cause death. Hemodialysis, the most common form of dialysis, generally requires a patient to visit a dialysis center at least three times per week for a three- to five-hour session, significantly reducing quality of life.
Phosphorus, a vital element required for most cellular processes, is present in almost every food in the Western diet, and, in individuals with normal kidney function, any excess dietary phosphorus is efficiently removed by the kidneys and excreted in urine. In adults with functioning kidneys, normal serum phosphorus levels are 2.5 to 4.5 mg/dL. With kidney failure, elevated phosphorus becomes harmful and is diagnosed as hyperphosphatemia when serum phosphorus levels are greater than 5.0 mg/dL. Although patients with ESRD rely on dialysis to eliminate harmful agents, phosphorus is not readily removed by the procedure and other means of managing phosphorus levels must be employed.
In ESRD patients, excess levels of phosphorus have been shown to lead to an increase in cardiovascular disease risk, as well as increases in serum FGF‑23, an important regulator of phosphate and vitamin D metabolism. Highly elevated levels of FGF23 is an independent risk factor for adverse cardiac clinical outcomes as well as the development of secondary hyperparathyroidism, or SHPT, marked by elevated parathyroid hormone. SHPT is associated with renal osteodystrophy, a condition of abnormal bone growth characterized by brittle bones.
Since dialysis is unable to efficiently eliminate excess phosphorus, ESRD patients are put on restrictive low phosphorus diets and are currently prescribed medications called phosphate binders, the only interventions currently marketed for the treatment of hyperphosphatemia. Phosphate binders act by binding dietary phosphorus and commonly need to be taken with meals and snacks. They include calcium, iron or lanthanum, a rare-earth metal, which bind to and precipitate with dietary phosphate in the GI tract. The goal of these phosphate binders is for patients to eliminate, through their stool, the precipitated phosphorus that comes from the food they ingest. Phosphate binders have a number of limitations, including:
- systemic excess absorption of calcium, iron or lanthanum, resulting in side effects and other unintended consequences for ESRD patients, and
- significant challenges with patient compliance because of the large quantity and/or mass of the binders that must be taken each day.
Safety and tolerability have also been significant concerns with many approved phosphate binders, with side effects that include long-term vascular calcification with calcium-based binders and iron-overload with iron-based binders. The more common side effects of certain approved phosphate binders include GI-related adverse events such as nausea, vomiting, diarrhea, and dyspepsia as well as hypercalcemia for calcium-based binders and discolored feces for iron-based binders.
ESRD patients, who generally are severely restricted in their fluid intake, are prescribed as many as 12 or more phosphate binder pills per day, among other medications. The amount of phosphorus a binder can remove is limited by its binding capacity, and therefore, increasing the dose, and hence the pill burden, of the binder is the only way to increase the amount of phosphorus being bound and excreted. As a result of pill burden and mass, as well as a number of side effects, prescribed phosphate binder doses are intolerable for many patients, leading to a lack of treatment adherence and compliance.
Ardelyx is developing tenapanor for the treatment of hyperphosphatemia in ESRD patients on dialysis, as the company believe it has the potential to address certain of the key limitations of current treatments and offer a completely new mechanism of action. If approved, tenapanor would be the first small molecule/non-binder approach to treating hyperphosphatemia, with a unique mechanism of action that acts by inhibiting, or blocking, the NHE3 transporter in the GI tract to reduce the absorption of dietary sodium. When tenapanor blocks the NHE3 sodium transporter in the GI tract, it reduces the absorption of dietary sodium, resulting in an increase in protons within the cells. This increase in protons causes a selective reduction in phosphate uptake by tightening junctions or pores that are involved in the regulation of phosphate homeostasis, which then limits the amount of dietary phosphate that can pass from the gut into the blood. Ardelyx has not observed this impact on other ions, nutrients or macromolecules in its clinical trials. Ardelyx has submitted a manuscript for publication of this mechanism in a scientific peer-reviewed journal.
This unique mechanism of action allows tenapanor to be active in many patients at a dose of 10mg to 30mg twice daily as opposed to the multiple gram quantities per day required of the phosphate binders. Over the course of a week, the amount of tenapanor required would be less than 500mg, or a total of 14 small pills, whereas the amount of phosphate binder required, based on package inserts, would be 10 to 30 grams, or up to 64 large pills, depending on the phosphate binder. The company believe this significant pill burden advantage will result in better adherence and compliance which could lead to more consistent efficacy in ESRD patients on dialysis.
Tenapanor has been specifically designed to work exclusively within the GI tract, thereby significantly reducing the amount of drug that is absorbed into the bloodstream and the potential side effects that could occur. In human studies of orally-administered tenapanor, the drug was detected in the blood in less than 1% in thousands of collected serum samples, and even in those, at very low levels (< 1.5 ng/mL). Ardelyx has evaluated tenapanor across 20 clinical studies in over 2,500 individuals to date.
Clinical data supporting tenapanor in hyperphosphatemia
In February 2017, the company announced data from its first Phase 3 clinical trial evaluating tenapanor for the treatment of hyperphosphatemia in ESRD patients on dialysis.
The Phase 3 trial was an eight-week, double-blind, randomized trial, with a four-week placebo-controlled randomized withdrawal period. The company enrolled a total of 219 ESRD patients with hyperphosphatemia who are on dialysis. Enrolled patients were randomized evenly into three arms, in which all groups received tenapanor for eight weeks.
Tenapanor was administered at doses of 3 mg or 10 mg twice-daily and in a dose-titration arm starting at 30 mg twice-daily with the option to down-titrate once a week during the first four weeks to 20, 15, 10 and 3 mg twice-daily, based on GI tolerability. After the end of the eight-week treatment period, patients were re-randomized 1:1 to either remain on their current tenapanor dose or switch to placebo for a four-week, placebo-controlled, randomized withdrawal period.
The primary endpoint of the trial was the difference in change in serum phosphorus between the pooled tenapanor-treated patients and placebo-treated patients from the end of the eight-week treatment period to the end of the four-week randomized withdrawal period, in the responder population. The responder population, which was reviewed by the FDA, is defined as patients who demonstrate a greater than or equal to 1.2 mg/dL decrease in serum phosphorus from baseline during the initial eight-week treatment period.
The study demonstrated a statistically significant difference in serum phosphorus levels from the end of the eight-week treatment period to the end of the four-week randomized withdrawal period between the tenapanor-treated group and the placebo-treated group in the responder patient population (mean -1.01 mg/dL, median of -1.3 mg/dL) and met its primary endpoint (95% confidence interval, -1.44, -0.21, LSmean -0.82 mg/dL, p=0.01). The responder population (n=80 out of 164) had a mean reduction in serum phosphorus from baseline to the end of the eight-week treatment period of 2.56 mg/dL, with a reduction of up to 5.7 mg/dL. Notably, in this group, 33% of patients had a reduction in serum phosphorus of greater than 3 mg/dL.
Tenapanor was well-tolerated in the trial. In the eight-week treatment period, the only adverse event that affected more than five percent of patients treated with tenapanor was diarrhea (39%), a patient-reported side effect of loosened stool or increased frequency in bowel movements regardless of magnitude. In the four-week randomized withdrawal period, there was a diarrhea rate of 1.2% for patients treated with tenapanor compared with 2.4% on placebo. Treatment discontinuations due to diarrhea for patients on tenapanor was 7.8% (n=17). There were no discontinuations due to diarrhea in the randomized withdrawal period.
In order to fully assess GI tolerability, patients used an eDiary to record the frequency of daily bowel habits, as well as stool form using the Bristol Stool Form Scale, or BSFS. During the eight-week treatment period, there was an average 0.4 per day increase in bowel movement frequency from baseline, and during the four-week randomized withdrawal period, there was an average 0.29 per day increase as compared to placebo. Average bowel movement frequency was within the normal range in all groups. During the eight-week treatment period, there was an average 0.87 point increase in BSFS from a baseline score of 4.2, out of a maximum of seven, where seven is liquid stool. During the four-week randomized withdrawal period, there was an average 0.7 point difference in BSFS between placebo (4.4) and tenapanor treatment (5.1).
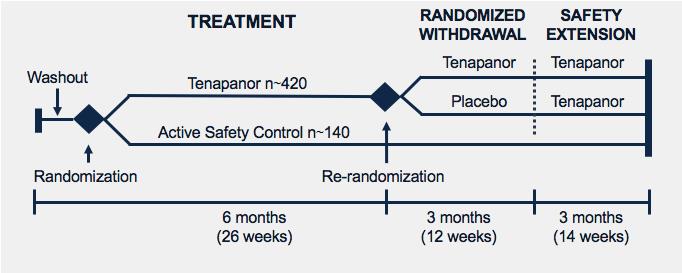
Ardelyx has initiated a second Phase 3 study of tenapanor for the treatment of hyperphosphatemia in ESRD patients on dialysis. The study's design, shown in the figure, will include a 26‑week open-label treatment period, with a 12-week randomized withdrawal period followed by an additional 14‑week safety extension. Results from this study are expected in 2019. The company currently intend to build its own sales and marketing organization to market and sell tenapanor for hyperphosphatemia in the United States.
The hyperphosphatemia market
Phosphate binders are the only drugs marketed for the treatment of hyperphosphatemia in ESRD patients. The various types of phosphate binders commercialized in the United States include the following:
- Calcium carbonate (many over-the-counter brands including Tums and Caltrate)
- Calcium acetate (several prescription brands including PhosLo and Phoslyra)
- Lanthanum carbonate (Fosrenol marketed by Shire)
- Sevelamer hydrochloride (Renagel, marketed by Sanofi)
- Sevelamer carbonate (Renvela, marketed by Sanofi)
- Sucroferric oxyhydroxide (Velphoro, marketed by Vifor Fresenius)
- Ferric citrate (Auryxia, marketed by Keryx)
The hydrochloride form of sevelamer, Renagel, was launched in the United States by Genzyme Corporation in 1998 prior to its acquisition by Sanofi, and the carbonate form, Renvela, was launched in 2008. Sanofi reported €922 million ($1.05 billion) in worldwide sales of sevelamer during 2016 and €802 million ($0.98 billion) in 2017. Generic sevelamer carbonate has been approved in certain jurisdictions in Europe since 2015 and in the U.S. market since June 2017.
In addition to the currently marketed phosphate binders, Ardelyx is aware of at least two other binders in development, including fermagate (Alpharen), an iron-based binder in Phase 3 studies being developed by Opko Health, Inc., and PT20, an iron-based binder in Phase 3 being developed by Shield Therapeutics.
According to the most recent data available from the U.S. Renal Data System, in 2015 there were 444,337 patients on hemodialysis in the United States. Additionally, according to the European ERA-EDTA Registry 2015 Annual Report and a study in 2014 by the Japanese Society for Dialysis Therapy, there were approximately 317,000 patients on hemodialysis in Europe and about 255,000 in Japan. The company estimate, based on phosphate binder utilization, the only approved therapies for hyperphosphatemia, that there are approximately 310,000, 250,000 and 260,000 ESRD patients with hyperphosphatemia in the United States, countries in Europe and Japan, respectively, resulting in approximately 820,000 ESRD patients with hyperphosphatemia in such countries.
Because many ESRD patients with hyperphosphatemia are unable to lower serum phosphorus levels to below 5.5 mg/dL with currently marketed phosphate binders, the company believe there is a significant medical need for new agents with new mechanisms, demonstrated efficacy, a strong safety profile, and significantly lower pill burden. The company believe that tenapanor, if approved, has the potential to have the lowest pill burden and mass among any currently marketed hyperphosphatemia products, with milligram rather than gram quantities. In addition, the company may evaluate whether tenapanor has the potential to be used in combination with phosphate binders for those patients who cannot achieve adequate phosphate control with a single agent.
The company's intention is to build a United States-focused, highly efficient, specialized sales and marketing organization focused on nephrology. The nephrology market is a concentrated market strongly influenced by key opinion leaders. There were 10,083 nephrologists in the United States in 2015 and 6,620 dialysis facilities in the United States that offer in-center dialysis. Based on the experience of its management team, the company believe that a specialty salesforce is appropriate for this marketplace. The company believe that tenapanor for the treatment of hyperphosphatemia could represent a market opportunity of between $500 million and $700 million in the United States.
As a first-in-class treatment, and as the first non-binder option with a well-tolerated safety profile, the company believe tenapanor could address the significant pill burden challenges and intolerability that patients experience with today’s binder treatments. After the second Phase 3 study readout, if successful, the company intend to submit a New Drug Application to the FDA in 2019 and would plan for a potential launch 2020.
To bring tenapanor to patients outside the United States, the company intend to establish strategic collaborations with industry leading pharmaceutical companies with established commercial infrastructures. In 2017, the company entered into two collaboration partnerships, and the revenues recorded from those collaboration partnerships in 2017 accounted for more than 10% of total revenues recorded during the year ended December 31, 2017.
License agreement with KHK
In November 2017, the company entered into a license agreement (“KHK License Agreement”) with KHK under which the company granted KHK an exclusive license to develop and commercialize tenapanor in Japan for the treatment of cardiorenal diseases and conditions, excluding cancer (“KHK Field”). The company retained the rights to tenapanor outside of Japan, and also retained the rights to tenapanor in Japan for indications other than those in the KHK Field. Pursuant to the KHK License Agreement, KHK is responsible for all of the development and commercialization costs for tenapanor in the KHK Field in Japan.
Under the KHK License Agreement, Ardelyx is responsible for supplying the tenapanor drug product for KHK’s use in development and during commercialization until KHK has assumed such responsibility. Additionally, Ardelyx is responsible for supplying the tenapanor drug substance for KHK’s use in development and commercialization throughout the term of the KHK License Agreement, provided that KHK may exercise an option to manufacture the tenapanor drug substance under certain conditions.
Under the terms of the KHK License Agreement, Ardelyx has received a $30.0 million upfront payment and are eligible to receive up to an additional $130.0 million in development and commercialization milestones, based upon currency exchange rates as of the effective date of KHK License Agreement.
Ardelyx is also eligible to receive royalties based on aggregate annual net sales of the licensed products at a high teen percentage, subject to certain single digit reductions under certain circumstances described in the KHK License Agreement.
The KHK License Agreement will continue until all of KHK’s applicable payment obligations under the KHK License Agreement have been performed or have expired, or the agreement is earlier terminated. Under the terms of the KHK License Agreement, the company and KHK each have the right to terminate the agreement for material breach by the other party. In addition, KHK may terminate the agreement for convenience; for certain safety reasons or if certain primary endpoints under an applicable development plan are not met despite KHK’s commercially reasonable efforts and KHK reasonably determines that it cannot obtain regulatory approval. KHK may also terminate the agreement if certain pivotal clinical trials conducted by it do not meet their primary endpoints. The company may terminate the KHK License Agreement if KHK challenges any patents licensed to KHK under the agreement.
License agreement with Fosun
In December 2017, the company entered into a license agreement (the “Fosun License Agreement”) with Fosun Pharma under which the company granted Fosun Pharma an exclusive license to develop and commercialize tenapanor in China for the treatment, diagnosis or prevention of ![]() irritable bowel syndrome with constipation and chronic idiopathic constipation, (ii) hyperphosphatemia related to chronic kidney disease and (iii) other diseases or conditions for which the company obtain marketing approval in either the US or China (collectively, “Fosun Field”). The Fosun Field excludes the treatment of cancer. The company retained the rights to tenapanor outside of China, and also retained the rights to tenapanor in China for indications other than those in the Field. Pursuant to the terms of the Fosun License Agreement, Fosun Pharma is responsible for all of the development and commercialization costs for tenapanor in the Fosun Field in China.
irritable bowel syndrome with constipation and chronic idiopathic constipation, (ii) hyperphosphatemia related to chronic kidney disease and (iii) other diseases or conditions for which the company obtain marketing approval in either the US or China (collectively, “Fosun Field”). The Fosun Field excludes the treatment of cancer. The company retained the rights to tenapanor outside of China, and also retained the rights to tenapanor in China for indications other than those in the Field. Pursuant to the terms of the Fosun License Agreement, Fosun Pharma is responsible for all of the development and commercialization costs for tenapanor in the Fosun Field in China.
Under the terms of the Fosun License Agreement, Ardelyx is responsible for supplying the tenapanor drug product for Fosun Pharma’s use in development and during commercialization until Fosun Pharma has assumed such responsibility. Additionally, Ardelyx is responsible for supplying the tenapanor drug substance for Fosun Pharma’s use in development and commercialization throughout the term of the Fosun License Agreement.
Under the terms of the Fosun License Agreement, the company received an upfront payment of $12 million and are eligible to receive additional milestones of up to $113 million in the aggregate, as well as tiered royalty payments on aggregate net sales ranging from the mid-teen percent to twenty percent, subject to certain reductions under certain circumstances, as described in the Fosun License Agreement.
The Fosun License Agreement will continue until all of Fosun Pharma’s applicable payment obligations under the License Agreement have been performed or have expired, or the agreement is earlier terminated. Under the terms of the Fosun License Agreement, the company and Fosun Pharma each have the right to terminate the agreement for material breach by the other party or in the event of insolvency by the other party. In addition, Fosun Pharma may terminate the agreement for convenience and the company may terminate the agreement if Fosun Pharma challenges any patents licensed to it under the agreement.
RDX013 Program: Small Molecule for Treating Hyperkalemia
RDX013 is its novel, small molecule program for the potential treatment of hyperkalemia. The company's RDX013 approach works by tapping into the GI tract’s natural ability to secrete potassium into the lumen of the gut to reduce serum potassium levels. This mechanism differs significantly from the potassium binders currently on or approaching the market. For a potassium binder to work, it must be present when dietary potassium is ingested so that the agent can bind the potassium and prevent its absorption in the gut. This results in the need for large quantities of binder in order to bind the large amounts of potassium in the diets of most individuals. In contrast, the company observed in its preclinical models that a small amount of RDX013 could cause potassium to be secreted into the lumen of the gut. In this way, the company believe that RDX013 may have the potential to lower serum potassium whether or not potassium is present in the diet and could result in a very low pill burden, potentially allowing better patient compliance, longer-term use and potentially better efficacy than potassium binders. As described below, certain medications commonly administered to patients with CKD and/or heart failure can also cause hyperkalemia. With the successful development of an effective potassium secretagogue to treat hyperkalemia in a small, convenient pill format, the company believe its RDX013 approach may allow nephrologists and cardiologists with an opportunity to treat hyperkalemia chronically without reducing the dose of these medications.
Hyperkalemia is generally defined as the presence of blood potassium levels greater than 5.0 mEq/L. Normal levels are 3.5 to 5.0 mEq/L. When hyperkalemia is severe, above 7.0 mEq/L, there is a significantly increased risk of death because of the potential for heart conductance problems.
Hyperkalemia can be caused by a variety of issues. Kidney disease can result in the elevation of potassium in the blood. Certain drugs such as the common hypertension medications known as RAAS inhibitors, which inhibit the renin-angiotensin-aldosterone system, can cause hyperkalemia. As a result, the dosage of RAAS inhibitors must often be significantly reduced in patients whose potassium levels are elevated (such as in those with CKD and heart failure). Because of the risk of hyperkalemia, several published guidelines have suggested that physicians should reduce and possibly discontinue RAAS inhibitors in order to manage the risk of hyperkalemia in CKD and heart failure patients. The alternative medications used to control hypertension, including diuretics and calcium channel blockers, are less effective than RAAS inhibitors, particularly in patients with failing kidneys and severe hypertension. According to the 2015 publication Market Dynamix: Hyperkalemia, released by Spherix Global Insights, U.S. cardiologists reported that of the patients who would benefit from RAAS inhibition, up to 38% of patients with heart failure and up to 55% of patients with both heart failure and CKD are being administered a sub-optimal dose or none at all. Nephrologists reported that at least one-third of patients who would benefit from RAAS inhibition receive a sub-optimal dose or none at all. The company believe there is clearly a strong medical need for new medications that control hyperkalemia in order to allow for optimal use of RAAS inhibitors to control hypertension in these patient populations.
The hyperkalemia market
Of the people with CKD and/or heart failure in the United States, the company estimate that there are approximately 2.1 million people who also have occurrences of hyperkalemia. According to a retrospective study conducted in 2005 of a national cohort of 246,000 patients cared for in the Veterans Health Administration, about 21% and 42% of patients with CKD Stage 3b and Stage 4, respectively, had a hyperkalemic event during a 12-month period, suggesting that hyperkalemia affects about 900,000 individuals with CKD Stage 3b or Stage 4 in the United States. According to the United States Renal Data System 2014 Atlas of CKD & ESRD, over 50% of CKD Stage 3b and Stage 4 patients are prescribed RAAS inhibitors to control hypertension and to slow the course of CKD.
Additionally, the number of adults in the U.S. living with heart failure is about 6.5 million, based on data collected in the National Health and Nutrition Examination Survey, which is taken in stages over multiple years. The company's proprietary research suggests that up to 16%, or approximately 1,000,000, of these patients had hyperkalemia during a 12-month period. Over half of heart failure patients are prescribed RAAS inhibitors. The company's proprietary research also suggests that up to 200,000 patients with ESRD in the U.S. could benefit from an agent that treats hyperkalemia.
Ardelyx is aware of at least two drugs approaching or on the market for the treatment of hyperkalemia. Veltassa (patiromer FOS), an oral, polymer-based potassium binder, was approved for marketing by the FDA in October 2015 and was commercially launched by Relypsa, which was acquired by Galenica AG for $1.5 billion in September 2016. However, according to a 2017 survey of more than 200 nephrologists and cardiologists, conducted by Spherix Global Insights, about half of those surveyed note that, even with Veltassa available, there remains a high unmet need for new treatments for hyperkalemia.
Additionally, ZS Pharma submitted an NDA in June 2015 for ZS-9, a sodium zirconium cyclosilicate-based oral potassium binder. ZS Pharma was acquired by AstraZeneca in December 2015 for $2.7 billion.
The company believe that, unlike these agents which require large amounts of drug for the desired effect, RDX013 may have the potential to lower serum potassium whether or not potassium is present in the diet and could result in very low pill burden, allowing better compliance, longer-term use and potentially better efficacy than potassium binders.
If Ardelyx is successful in developing RDX013 and obtaining marketing authorization from the FDA, the company would expect to leverage the renal sales and marketing organization that the company intend to build to support commercialization in the United States of tenapanor for treating hyperphosphatemia in dialysis patients.
Tenapanor: NHE3 Inhibitor for Treating IBS-C
In addition to its development for hyperphosphatemia, Ardelyx has completed development of tenapanor for the treatment of IBS-C and are pursuing strategic collaborations to bring it to market in this indication. Ardelyx has completed two Phase 3 clinical trials in patients with IBS-C (T3MPO-1 and T3MPO-2) along with a long-term safety study (T3MPO-3) and expect to submit an NDA to the FDA in the second half of 2018.
IBS-C is a GI disorder in which abdominal pain or discomfort is associated with constipation, and which significantly impacts the health and quality of life of affected patients. A study published in the American Journal of Gastroenterology in 2015, showed that over 50% of IBS-C patients rated their pain, constipation and straining as being “extremely bothersome.” In the same study, GI symptoms led to an average 4.9 days of “disrupted productivity” and 0.8 days of missed work per month. There is no specific test or biomarker for IBS-C and therefore its presence is diagnosed by symptoms and by eliminating other disorders. IBS-C is very similar to chronic constipation and is clinically distinguished by a significant abdominal pain component.
Tenapanor is a minimally-systemic small molecule that acts locally in the GI tract to inhibit the sodium transporter NHE3 and reduce sodium uptake from the gut. Part of its mechanism to treat constipation caused by IBS-C is an osmotic effect in the intestines – water follows salt and stool is gently loosened by the body’s own fluids. In addition to this mechanism, data from nonclinical studies, which were presented in November 2017 at the American Society of Nephrology Annual Meeting, suggest that tenapanor reduced abdominal pain caused by IBS-C through the inhibition of TRPV-1 dependent signaling. TRPV-1, better known as the "hot chili pepper receptor," is a well-established pain target known for transmitting painful stimuli from a variety of sources including heat, protons and inflammatory molecules. Using an established rodent model of IBS-like colonic hypersensitivity, preclinical data showed that tenapanor treatment reduced visceral hypersensitivity (pain in the internal organs) and normalized colonic sensory neuronal excitability and TRPV-1 currents. Treatment with tenapanor also increased stool excretion and stool water content. In these nonclinical studies, tenapanor had a superior effect on visceral hypersensitivity than placebo or PEG, a well-known laxative not known to have an analgesic effect.
Phase 3 clinical data supporting tenapanor in IBS-C
At the end of 2017, the company completed its Phase 3 program, the T3MPO program, for tenapanor for the treatment of IBS-C. This included two Phase 3 studies, T3MPO-1 and T3MPO-2, and a long-term safety extension study, T3MPO-3. In the discussion below, statistical significance is denoted by p-values. The p-value is the probability that the reported result was achieved purely by chance (e.g., a p-value <0.001 means that there is a less than a 0.1% probability that the observed change was purely due to chance). Generally, a p-value less than 0.05 is considered statistically significant.
In May 2017, the company reported data from the first Phase 3 study, T3MPO-1. T3MPO-1 was a 12-week, double-blind, placebo-controlled, multi-center, randomized trial with a four-week, randomized withdrawal period conducted in a total of 610 patients meeting the ROME III criteria for the diagnosis of IBS-C. Patients were randomized one to one to receive either 50 mg of tenapanor or placebo twice-daily. The trial included a two-week screening period, during which patients with active disease, based on bowel movement frequency and abdominal pain score recorded in a daily phone diary, were randomized into the trial.
The study achieved statistical significance for the primary endpoint and seven of eight secondary endpoints. The primary endpoint, the combined responder rate for six of 12 weeks, showed that a greater proportion of tenapanor-treated patients compared to placebo-treated patients (27.0% vs 18.7%, p=0.02) had at least a 30% reduction in abdominal pain and an increase of one or more complete spontaneous bowel movements, or CSBM, in the same week for at least six of the 12 weeks of the treatment period. The CSBM responder rate in the six of 12 weeks (defined as having an increase of at least one CSBM from baseline during a week for six of 12 weeks) did not reach statistical significance (33.9% vs. 29.4%, p=0.27); however, the abdominal pain responder rate in the six of 12 weeks (defined as having at least a 30% decrease in abdominal pain from baseline during a week for six of 12 weeks) did achieve statistical significance (44.0% vs. 33.1%, p=0.008). In the nine of 12 treatment weeks, the study achieved statistical significance for the combined responder rate (13.7% vs. 3.3%, p<0.001), in which patients must have had an increase of at least one CSBM from baseline and at least three CSBMs during a week for nine of 12 weeks as well as a 30% or greater reduction in abdominal pain for nine of the 12 weeks. In addition, for the nine of 12 weeks, the study achieved statistical significance for the individual CSBM responder rate (16.9% vs. 5%, p<0.001) and the abdominal pain responder rate (30.3% vs. 19.4%, p=0.003).
Tenapanor was well-tolerated in the study. The only adverse events observed in more than two percent of patients treated with tenapanor, as compared with placebo, were diarrhea (14.6% vs 1.7%) and nausea (2.6% vs 1.7%). The placebo adjusted discontinuation rate due to diarrhea was 5.3%.
In October 2017, the company reported results from the second Phase 3 study of tenapanor for the treatment of IBS-C, T3MPO-2, a 26-week, double-blind, placebo-controlled, multi-center, randomized trial in 593 patients. Patients were randomized one to one to receive either 50 mg of tenapanor or placebo twice-daily. The trial included a two-week screening period, during which patients with active disease, based on bowel movement frequency and abdominal pain score recorded in a daily phone diary, were randomized into the trial.
The study achieved statistical significance for the primary endpoint and all secondary endpoints evaluated for the topline results and demonstrated the ability to normalize bowel movements. The primary endpoint, the combined responder rate for six of 12 weeks, showed that a greater proportion of tenapanor-treated patients compared to placebo-treated patients (36.5% vs. 23.7%, p<0.001) had at least a 30% reduction in abdominal pain and an increase of one or more CSBMs in the same week for at least six of the 12 weeks of the treatment period. The study achieved statistical significance for the CSBM and abdominal pain responder rates in the six of 12 weeks (47.4% vs. 33.3%, p<0.001; 49.8% vs. 38.3%, p=0.004). In the nine of 12 treatment weeks, the study achieved statistical significance for the combined responder rate (18.4% vs. 5.3%, p<0.001), in which patients must have had an increase of at least one bowel movement from baseline and at least three per week and a 30% reduction in abdominal pain for nine of the 12 weeks. In addition, for the nine of 12 weeks, the study achieved statistical significant for the individual CSBM responder rate (22.2% vs. 6.0%, p<0.001) and the abdominal pain responder rate (35.8% vs. 26.7%, p=0.015). The study also achieved statistical significance for all three durable responder endpoints, the combined responder rate (18.1% vs. 5.0%, p<0.001), CSBM responder rate (21.2% vs. 5.7%, p<0.001) and abdominal pain responder rate (34.8% vs. 26.7%, p=0.028), which are identical to the nine of 12-week responder endpoints, except the response must also occur in three of the last four treatment period weeks.
Tenapanor was well-tolerated in the T3MPO-2 study. The only adverse events observed in more than two percent of patients in the tenapanor-treated group that were also greater than placebo were diarrhea (16.0% vs. 3.7%), flatulence (3.1% vs. 1.0%), nasopharyngitis (4.4% vs. 3.7%) and abdominal distension (3.4% vs. 0.3%). The placebo adjusted discontinuation rate due to diarrhea was 5.8%.
Patients who completed T3MPO-1 and T3MPO-2 were eligible to enter T3MPO-3, its open-label, long-term safety trial where patients could continue to receive tenapanor for up to one year. In late 2017, the company completed T3MPO-3 and have reported results that tenapanor had a mean compliance rate of approximately 98%, and that tenapanor was well-tolerated among the 240 patients treated. Of patients treated, 9.2% reported experiencing diarrhea, with 1.7% of patients discontinuing treatment due to diarrhea. The overall discontinuation rate in the study was 2.1%.
Based on the positive results from the T3MPO program, Ardelyx is preparing to submit an NDA to the FDA to request marketing approval of tenapanor for the treatment of IBS-C in the United States.
To support the global commercialization of tenapanor for IBS-C, Ardelyx is pursuing strategic collaborations to bring tenapanor to patients. In December 2017, the company entered into a license agreement to provide Fosun Pharma with the exclusive rights to develop and commercialize tenapanor in China for the treatment of patients with IBS-C and hyperphosphatemia related to chronic kidney disease.
The IBS-C market
Numerous treatments exist for the constipation component of IBS-C, many of which are over-the-counter. There are three prescription products marketed for IBS-C: ![]() Linzess (linaclotide), marketed by Ironwood Pharmaceuticals and Allergan, (ii) Amitiza (lubiprostone), marketed by Takeda and Sucampo, a wholly-owned subsidiary of Mallinckrodt, and (iii) Trulance (plecanatide), marketed by Synergy Pharmaceuticals.
Linzess (linaclotide), marketed by Ironwood Pharmaceuticals and Allergan, (ii) Amitiza (lubiprostone), marketed by Takeda and Sucampo, a wholly-owned subsidiary of Mallinckrodt, and (iii) Trulance (plecanatide), marketed by Synergy Pharmaceuticals.
The company believe that tenapanor may offer a significant benefit over currently marketed drugs like Amitiza, Linzess and Trulance in part because of the profile demonstrated in its Phase 3 program, which showed best-in-class efficacy results for the 9-of-12 week combined responder rates. Within the United States, there are approximately 11 million patients that suffer that suffer from IBS-C. There is significant unmet need for prescription medications, where, according a 2015 American Gastroenterological Association report, only 1 in 4 treated patients are very satisfied with the current FDA approved treatments in IBS-C.
Other GI Programs
In addition to tenapanor for IBS-C, Ardelyx has an early GI pipeline comprised of RDX8940, a minimally absorbed, oral TGR5 agonist for which the company submitted an investigational new drug application, or IND, in late 2016; RDX011, its second-generation NHE3 inhibitor; and its RDX023 program for the development of gut-biased farnesoid X receptor, or FXR, agonists. While Ardelyx is not currently actively developing these programs, they represent potential collaboration opportunities to support their continued development.
Intellectual Property
The company's commercial success depends in part on its ability to obtain and maintain proprietary protection for its drug candidates, manufacturing and process discoveries, and other know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing its proprietary rights. The company's policy is to seek to protect its intellectual property by, among other methods, filing U.S. and foreign patent applications related to its proprietary technology and inventions that are important to the development and operation of its business. The company also rely on trade secrets and careful monitoring of its proprietary information to protect aspects of its business that are not amenable to, or that the company do not consider appropriate for, patent protection.
The patent positions of biopharmaceutical companies like it are generally uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Consequently, the company do not know whether any of its product candidates will be protectable or remain protected by enforceable patents. The company cannot predict whether the patent applications Ardelyx is currently pursuing will issue as patents in any particular jurisdiction or whether the claims of its issued patents will provide sufficient proprietary protection from competitors. Any patents that the company hold may be challenged, circumvented or invalidated by third parties. If third parties prepare and file patent applications in the United States that also claim technology or therapeutics to which Ardelyx has rights, the company may have to participate in interference proceedings in the U.S. Patent and Trademark Office, or USPTO, to determine priority of invention, which would result in substantial costs to it even if the eventual outcome is favorable to it.
The term of individual patents depends upon the legal term of the patents in countries in which they are obtained. In most countries, including the United States, the patent term is generally 20 years from the earliest date of filing a non-provisional patent application in the applicable country. In the United States, a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date.
In addition, in the United States, the Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of a U.S. patent as partial compensation for the patent term lost during the FDA regulatory review process occurring while the patent is in force. A patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to each regulatory review period may be extended and only those claims covering the approved drug, a method for using it or a method for manufacturing it may be extended. Similar provisions are available in the European Union and certain other foreign jurisdictions to extend the term of a patent that covers an approved drug.
The company may rely, in some circumstances, on trade secrets to protect its technology. Although the company take steps to protect its proprietary information and trade secrets, including through contractual means with its employees and consultants, third parties may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to its trade secrets or disclose its technology. Thus, the company may not be able to meaningfully protect its trade secrets. It is its policy to require its employees, consultants, outside scientific collaboration partners, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with it. These agreements provide that all confidential information concerning the business or financial affairs developed or made known to the individual during the course of the individual’s relationship with it is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual, and which are related to its current or planned business or research and development or made during the normal working hours, on its premises or using its equipment or proprietary information, are its exclusive property.
NHE3 patents
The company's NHE3 patent portfolio is wholly owned by it. This portfolio includes four issued U.S. patents, two issued Japanese patents, two issued Korean patents and one issued patent in each of the following territories: Australia, China, the European Union, Mexico, and Israel. These issued patents cover the composition and methods of using tenapanor and are predicted, without extension or adjustment, to expire in 2029. Ardelyx has related national patent applications pending in Europe, China, India, Israel and a number of other countries. Any patents issuing from these patent applications are also predicted without extension or adjustment to expire in 2029.
Additional U.S. and international patent applications are pending covering additional methods of using tenapanor, and composition of matter and methods of using compounds that the company believe may be follow on compounds to tenapanor.
Other program patents
Ardelyx has patent applications pending in the United States and internationally that cover the compositions and methods of using its TGR5 agonists, its FXR agonists and compounds in its RDX013 program.
Manufacturing
To date, Ardelyx has relied upon third-party contract manufacturing organizations, or CMOs, to manufacture both the active pharmaceutical ingredient and final drug product dosage forms of its potential drug candidates used as clinical trial material. The company expect that the company will continue to rely upon CMOs for the manufacture of its clinical trial materials and for its commercial product requirements, when and if regulatory approval is received. The company's license agreements with KHK and Fosun Pharma require it to supply active pharmaceutical ingredient and final drug product dosage forms of tenapanor for their use in the development of tenapanor in each of their respective territories, and Ardelyx is further obligated to continue to supply active pharmaceutical ingredient to support their commercialization of tenapanor in each of their territories. The company expect that the company will use CMOs to satisfy its supply obligations to its collaboration partners.
Employees
As of December 31, 2017, the company had 75 full-time employees, including a total of 15 employees with Ph.D. degrees. Within its workforce, 54 employees are engaged in research and development and the remaining 21 in general management and administration, including finance, legal, and business development. None of its employees are represented by labor unions or covered by collective bargaining agreements. The company believe that the company maintain good relations with its employees.
Research and Development
The company's research and development costs were $75.5 million, $94.2 million and $39.9 million in the years ended December 31, 2017, 2016 and 2015, respectivel




