Arena Pharmaceuticals
Overview
Arena Pharmaceuticals (ARNA) is a biopharmaceutical company focused on developing novel, small molecule drugs with optimized receptor pharmacology designed to deliver clinical utility across therapeutic areas. The company's proprietary, internally-developed pipeline includes multiple potentially first- or best-in-class programs.
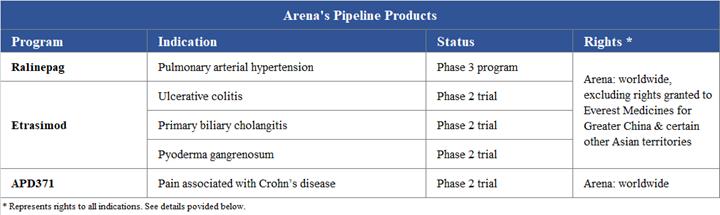
The company's three most advanced investigational clinical programs are: ralinepag (APD811), which Arena Pharmaceuticals is currently preparing for a Phase 3 program for pulmonary arterial hypertension, or PAH; etrasimod (APD334), which is being studied in Phase 2 trials for immune and inflammatory conditions with an initial focus on ulcerative colitis and hepatic conditions; and APD371 for visceral pain conditions and which is being studied in a Phase 2 trial for treatment of pain associated with Crohn's disease, or CD.
In addition, Arena Pharmaceuticals has collaborations with the following pharmaceutical companies: Everest Medicines Limited (ralinepag and etrasimod in Greater China and select countries in Asia), Axovant Sciences GmbH (nelotanserin - Phase 2), Boehringer Ingelheim International GmbH (undisclosed target - preclinical), and Eisai Co., Ltd. and Eisai Inc., collectively, Eisai (BELVIQ®/BELVIQ XR® - marketed products).
Strategy
The primary elements of its strategic focus are to:
- Develop ralinepag – a next-generation, potent, highly selective oral IP receptor agonist intended for the treatment of PAH
- Develop etrasimod – a modulator of the sphingosine 1-phosphate, or S1P, receptor intended for the treatment of a broad range of immune and inflammatory conditions
- Develop APD371 – an agonist of the cannabinoid-2, or CB2, receptor intended for the treatment of a range of visceral pain conditions
- Develop its pipeline by efficiently managing its cash and development timelines, which may include entering strategic collaborations for certain clinical and preclinical programs
- Progress additional pipeline programs over time in select therapeutic areas
- Build a streamlined, high-performing and high-energy organization
Arena Pharmaceuticals, Inc., incorporated in the state of Delaware in April 1997, is located in San Diego, California. The company's development operations are located in San Diego and Zug, Switzerland. The company also have manufacturing operations in Zofingen, Switzerland, which are subject to a pending sale under an asset purchase agreement.
Pipeline of Development Programs and Commercial Products
Below is a summary of its internally developed, proprietary portfolio:
In addition, Arena Pharmaceuticals has several other partnered programs. The company discovered and developed lorcaserin (BELVIQ and BELVIQ XR), which is currently marketed by Eisai or its distributors in the United States, or US, and certain other countries for weight loss. Lorcaserin is also being tested in an ongoing cardiovascular outcomes trial, or CVOT, for the reduction of major cardiovascular events and progression to type 2 diabetes. The company also discovered nelotanserin which is partnered with Axovant Sciences and currently in Phase 2 development for various neurologic disorders. The company also maintain a preclinical collaboration with Boehringer Ingelheim for undisclosed orphan targets in central nervous system disorders.
The company also own and have rights to other clinical and preclinical stage compounds that were internally discovered by it.
Ralinepag Program
Ralinepag is a next, generation potent, highly selective oral IP receptor agonist intended for the treatment of pulmonary arterial hypertension (PAH). Ralinepag was designed by it to deliver intravenous prostacyclin-like potency and pharmacokinetics in an oral tablet. In non-clinical experiments, ralinepag demonstrated potentially best-in-class activation of the IP receptor resulting in vasodilation, inhibition of smooth muscle cell proliferation and inhibition of platelet aggregation. Additionally, early stage studies of ralinepag pharmacokinetics in humans revealed an approximately 24-hour half-life and a low peak-to-trough ratio supporting therapeutic blood levels with once daily dosing.
In September 2014, ralinepag was granted orphan drug status for the treatment of PAH by the US Food and Drug Administration, or FDA.
PAH is a progressive, life-threatening disorder characterized by increased pressure in the pulmonary arteries that carry blood from the heart to the lungs. PAH occurs when the pulmonary arteries thicken or grow rigid. This makes blood flow more difficult. The heart must work harder to push blood through the arteries, and the arteries are unable to carry adequate blood to the lungs. The increased pressure strains the heart, which can limit physical activity, result in heart failure and reduce life expectancy. PAH will continue to worsen over time, even with proper treatment. Based on data from the Registry to EValuate Early And Long-term PAH disease management, or REVEAL, of patients in the US, there is an estimated five-year survival rate of 57% from diagnosis. The reported prevalence of PAH varies widely. One estimate is 15-50 cases/million with a higher female preponderance (approximately 3:1). More recently, the prevalence of PAH in the US among the privately insured (under age 65) and Medicare (age 65 and over) populations was estimated using administrative claims data in accordance with the current clinical classification of PAH. This analysis suggests PAH prevalence was 109 (71–146) cases/million among the under age 65 population, and 451 (384–519) cases/million for age 65 and over population or Medicare patients. Another estimate is that PAH affects about 500,000 individuals worldwide. A recent report characterizes the global market sales of PAH therapies as $5.8 billion in 2015, which are expected to increase to $6.7 billion by 2025.
PAH involves several interrelated mechanisms, with prostacyclin and thromboxane A2 playing a major role in maintaining pulmonary vascular tone through their balanced activity. Prostacyclin, released by endothelial cells, promotes vasodilation and inhibits platelet aggregation. Prostacyclin also has antiproliferative effects on vascular smooth muscle. Despite treatment guidelines, targeting the prostacyclin pathway has been primarily reserved for patients with advanced disease due to limitations of currently available options including parenteral prostacyclins which are the only PAH treatment that have demonstrated a mortality benefit.
Ralinepag Development
Arena Pharmaceuticals is currently preparing for a Phase 3 program for ralinepag.
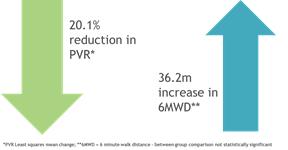
In 2017, the company announced topline results from a 22-week, randomized, double-blind, placebo-controlled Phase 2 trial evaluating the effectiveness in reducing pulmonary vascular resistance, or PVR, improving exercise capacity, tolerability and safety of ralinepag. In this trial, 40 patients with PAH received ralinepag and 21 received placebo. Topline results showed statistically significant improvement of both absolute and percentage change from baseline in PVR. Ralinepag also demonstrated numerical improvement in six-minute walk distance, or 6MWD, but as the study was not powered to show a difference in 6MWD from placebo, this was a not a statistically-significant finding. The safety and tolerability profiles were in line with other oral prostacyclins.
In 2013, we announced topline results from a multiple-dose, randomized, double-blind and placebo-controlled Phase 1 clinical trial evaluating multiple-ascending doses of ralinepag in healthy volunteers. In this trial, 40 healthy volunteers received ralinepag and 15 received placebo. The safety profile of ralinepag in this trial was characteristic of IP receptor agonists: the most frequent treatment-emergent adverse events were headache, nausea and jaw pain. One serious adverse event, transient atrial fibrillation, occurred in a single subject, and the study investigator considered it to be possibly treatment related. Further review revealed that the subject had multiple characteristics predisposing the patient to atrial fibrillation, including cardiac abnormalities prior to study start.
In 2011, we announced topline results of a Phase 1 clinical trial to evaluate the safety, tolerability and pharmacokinetics of single-ascending doses of ralinepag. The randomized, double-blind and placebo-controlled trial evaluated 32 healthy volunteers in four cohorts of eight participants each, with six randomized to ralinepag and two to placebo. Ralinepag was rapidly absorbed and demonstrated dose-proportional pharmacokinetic exposure over the tested dose range. Consistent with the expected pharmacology of ralinepag, the most common adverse events were headache, vomiting, nausea, jaw pain and flushing.
Ralinepag intellectual property
As of March 2, 2018, we owned issued patents covering compositions of matter for ralinepag and related compounds and methods of treatment utilizing ralinepag and related compounds, synthetic routes, and various solid state forms of ralinepag in 61 jurisdictions, including the United States, China, Japan, Germany, France, Italy, the United Kingdom, Spain, Canada, Russia, South Korea and Australia, and we had applications pending in three other jurisdictions, of which the ones with the largest pharmaceutical markets were Brazil and India. The patent on ralinepag issued by the US Patent and Trademark Office has serial number US 8,895,776, while the corresponding patent granted by the European Patent Office has serial number EP 2280696 B2. We also own issued patents and/or pending applications directed to synthetic processes and dosage regimens of ralinepag. The earliest priority date for the patents on ralinepag is 2008. The terms of these patents are capable of continuing into 2029 in most jurisdictions without taking into account any patent term adjustment or extension regimes of any country or any additional term of exclusivity we might obtain by virtue of the later filed patent applications.
Etrasimod Program
Etrasimod (APD334), is an oral, next generation, selective sphingosine 1-phosphate, or S1P, receptor modulator, discovered by Arena, designed to provide systemic and local cell modulation by selectively targeting S1P receptor subtypes 1, 4 and 5, while avoiding subtypes 2 and 3. In early stage studies, etrasimod exhibited potentially best-in-class pharmacokinetics and pharmacodynamics with rapid onset of action and rapid recovery of T lymphocytes. Selective binding with S1P receptor subtype 1 is believed to inhibit a specific subset of activated lymphocytes from migrating to sites of inflammation. The result is a reduction of circulating T and B lymphocytes that leads to anti-inflammatory activity and immune surveillance is maintained. The receptor subtypes 4 and 5 exhibit similar activity on additional proliferating immune cell types. Optimized pharmacology and pharmacokinetics may allow improved clinical utility across a broad range of immune-inflammatory conditions.
Ulcerative Colitis
Inflammatory bowel diseases, or IBD, like ulcerative colitis, or UC, and Crohn’s disease, or CD, are chronic inflammatory conditions of the gastrointestinal tract that affect approximately 1.6 million people in the US alone. The prevalence of UC and CD in the US are currently estimated at 914,000 and 684,000 patients, respectively. The prevalence of IBD in European Union, or EU, is estimated at 2.6 million with 1.1 million persons with CD and 1.5 million persons with UC. Both conditions have a significant impact on the patient’s quality of life and can in many cases be very aggressive and disabling.
UC is characterized by mucosal inflammation limited to the colon which involves the rectum in about 95% of cases and may extend to involve parts or all of the large intestine. In contrast, CD is characterized by full thickness inflammation that can occur anywhere in the gastrointestinal, or GI, tract but most typically involves the terminal ileum and colon; and causes fistulation and scarring. Symptoms for UC and CD can vary, depending on the location and severity of inflammation, but some of the most common are diarrhea, abdominal cramps, and rectal bleeding.
Important goals of therapy for UC are to induce and maintain remission while improving the patient’s quality of life. Currently available treatment options have limitations in terms of long-term efficacy and side effects, have complicated administration regimens, and often fail to induce or maintain remission. Therefore, we believe a significant unmet need remains for differentiated oral agents that are efficacious for induction and maintenance therapy with a favorable side effect profile. We believe that the oral once-daily dosing, selectivity, mechanism of action, and emerging clinical profile of etrasimod may represent a significant opportunity to provide patients with an effective treatment for UC with an improved safety and dosing profile over current therapies.
Primary Biliary Cholangitis
Primary biliary cholangitis, or PBC (previously referred to as primary biliary cirrhosis), is a chronic liver disease which is classified as a rare disease. The prevalence in the US is approximately 40 cases per 100,000 inhabitants. The incidence and prevalence of PBC in European countries are similar to those seen in the US.
Progressive bile-duct injury from portal and periportal inflammation could result in progressive fibrosis, cholangitis and eventually cirrhosis. Evidence to date suggests that immunological and genetic factors might cause the disease. Current treatment attempts to slow the progression rate of the disease and to alleviate the symptoms. Currently liver transplantation appears to be the only life-saving procedure for PBC patients.
Inflammation, the underlying cause of PBC, is believed to be T lymphocyte mediated. In research models with etrasimod, we have demonstrated modulation of the specific subtypes of T lymphocytes believed to be implicated in PBC.
Pyoderma Gangrenosum
Pyoderma gangrenosum, or PG, is a rare inflammatory skin disease characterized by painful recurrent ulcerations. Lesions may occur either in the absence of any apparent underlying disorder or in association with other diseases, such as UC, CD, and other conditions. The clinical course can be mild or malignant, and chronic or relapsing.
The etiology of PG has not yet been clearly determined, although it is suspected to be an autoimmune disease caused by dysregulation of the immune system. Approximately 50% of cases of PG are associated with other disorders, especially UC or CD.
Based upon the US Department of Health and Human Services' National Institutes of Health's Office of Rare Disease Research, the incidence of PG each year in the US has been estimated to be 1 person per 100,000 people.
Treatment is challenging, and the prognosis of PG remains unpredictable. Current treatments involve wound care and the use of anti-inflammatory agents, including antibiotics, corticosteroids, immune-suppressants and biologics, and attempts to target a broad spectrum of immunologic mediators and inflammatory cells, including T lymphocytes shown to be involved in PG. We believe reduction of lymphocytes by S1P receptor modulators such as etrasimod may represent a novel therapeutic approach in PG.
Etrasimod Development
Ulcerative Colitis
We are conducting a dose finding 12-week randomized, double-blind, placebo-controlled multinational Phase 2 clinical trial of etrasimod in moderate to severe UC. The aim of the trial includes investigating a clear dose response and establishing a clinically meaningful signal for the active arm(s) from placebo. The trial is expected to evaluate the effects of etrasimod at 1mg and 2mg versus placebo on multiple efficacy measures including a three-component partial Mayo Clinic Score, clinical remission, clinical response, and endoscopic improvement in up to 160 patients. Patients from this study have the possibility to continue after the initial 12-week study in an open-label extension study for up to 46 weeks with the focus on safety and maintenance of therapeutic effect.
Primary Biliary Cholangitis
We are conducting a 24-week open-label multinational Phase 2 trial of etrasimod in PBC. The aim of the study is to evaluate the safety and tolerability of etrasimod in this patient population as well as evaluate the effect of etrasimod on alkaline phosphatase, a key diagnostic marker of PBC, as well as other laboratory parameters.
Pyoderma Gangrenosum
We are conducting a Phase 2, proof of concept, open-label study to evaluate the efficacy and safety of etrasimod in patients with PG. The objective is to evaluate the efficacy, safety and tolerability of etrasimod in patients with PG over a 12-week treatment period. The study includes patients with diagnosed PG independent of IBD as a background disease.
Dermatologic Extraintestinal Manifestations of IBD
In March 2017, we initiated a Phase 2, proof of concept, open-label study evaluating the efficacy and safety of etrasimod in IBD patients with active dermatologic extraintestinal manifestations, or EIM. In November 2017, we announced that we decided to conclude the study since the UC study also includes patients with dermatological manifestations. We intend to assess the impact of etrasimod on EIMs of IBD in a combined data set from the two trials.
Prior Development
In January 2015, we announced top-line results from a Phase 1b multiple-ascending dose clinical trial for etrasimod. In the trial, etrasimod demonstrated a dose-dependent effect on lymphocyte count lowering in blood, with mean decreases from baseline of up to 69%. Lymphocyte counts, on average, recovered to baseline within one week of conclusion of dosing. There was a modest impact on heart rate, but none of the changes were classified by the investigator as clinically significant. There were also no findings with respect to pulmonary function or liver enzyme tests that were classified by the investigator as clinically significant. The most common treatment-emergent adverse events were mild or moderate contact dermatitis, headache, constipation and diarrhea, with none being clearly drug related. There were no discontinuations for adverse events, and no serious adverse events were observed.
The randomized, double-blind, placebo-controlled Phase 1b clinical trial evaluated the safety, tolerability, pharmacodynamics and pharmacokinetics of multiple-ascending doses of etrasimod. In five different dosing cohorts, 50 healthy volunteers received etrasimod and 10 healthy volunteers received placebo for 21 days.
Prior to commencing the Phase 1b multiple-ascending dose clinical trial for etrasimod, we completed a Phase 1 single-ascending dose clinical trial of the compound. This randomized, double-blind and placebo-controlled trial evaluated the safety, tolerability and pharmacokinetics of single-ascending doses of etrasimod in 40 healthy adult volunteers. In the trial, etrasimod demonstrated favorable pharmacokinetic and pharmacodynamic effects, a dose-responsive reduction in blood lymphocyte count and a slowing of heart rate that appears comparable to other S1P receptor modulators. The terminal half-life was approximately 35 hours.
Etrasimod intellectual property
As of March 2, 2018, we owned issued patents that cover compositions of matter for etrasimod and related compounds, methods of treatment utilizing etrasimod and related compounds, and various salts of etrasimod and crystalline forms thereof in 61 jurisdictions, including the United States, China, Japan, Germany, France, Italy, the United Kingdom, Spain, Canada, India, Russia, South Korea and Australia, and had applications pending in one other jurisdiction (Brazil). The patents on etrasimod issued by the US Patent and Trademark Office have serial numbers US 8,580,841 and US 9,126,932, while the corresponding patent granted by the European Patent Office has serial number EP 2326621 B2. We also own issued patents and/or pending applications directed to solid-state forms of etrasimod, dosage regimens for etrasimod and synthetic routes and intermediates useful in the manufacturing of etrasimod. The earliest priority date for the patents on etrasimod is 2008. The terms of these patents are capable of continuing into 2029 in most jurisdictions without taking into account any patent term adjustment or extension regimes of any country or any additional term of exclusivity we might obtain by virtue of the later filed patent applications.
APD371 Program
APD371, an orally available, potent, peripherally restricted, highly selective, full agonist of the CB2 receptor, is an internally discovered investigational drug candidate we are exploring for the treatment of visceral pain, specifically pain associated with CD.
Visceral pain is defined as pain that originates within muscle, pleura, connective tissue, nervous system or solid organs within the abdomen or peritoneum. It is distinct from somatic or neuropathic pain, and is perceived as stretching, pulling and distention, rather than by cutting, crushing, or burning more commonly associated with neuropathic pain. Visceral pain is one of the most common types of pain. For example, abdominal pain affects approximately 20% of the general population. Visceral pain may be caused by a diverse set of organic causes, such as inflammation (e.g., IBD (including CD and UC), pancreatitis, prostatitis, and vaginitis), obstruction (e.g., bowel obstruction, and nephrolithiasis), ischemia, and malignancy, among others. Visceral pain may also be caused by functional disorders such as interstitial cystitis, dyspepsia, irritable bowel syndrome (IBS), and vulvodynia.
A specific type of visceral pain, pain associated with CD, affects a significant portion of patients with underlying CD. CD affects approximately 684,000 patients in the US, and 20% of patients suffer from residual pain even while in remission.
Common treatments for visceral pain range from non-invasive, conservative approaches (e.g., physical therapy or acupuncture), to pharmacologic (e.g., tricyclic antidepressants acting as neurotransmitter reuptake inhibitors), and invasive interventions (e.g., bowel resection). Potent analgesics, such as opioids, can adversely affect GI function. Other commonly prescribed analgesics are often not potent enough and may lead to other GI side effects such as bleeding. Apart from linaclotide and lubiprostone, prescribed for IBS, no visceral-specific analgesics are available. Approximately one in eight CD patients is chronically treated with opioids.
The CB2 receptor is expressed in the GI nervous system, and in many tissues and organs of the abdomen. CB2 receptors are found peripherally on immune cells but also on microglia, terminal neurons, dorsal root ganglia, and on visceral sensory neurons. We believe selectively targeting the CB2 receptor may provide therapeutic benefit for visceral pain without the potential for dependence, abuse, and GI and cardiovascular side effects associated with opiates or nonsteroidal anti-inflammatory drugs, or NSAIDs, which are among the most common pain relievers. In addition to analgesic effects, APD371 may have anti-inflammatory properties.
APD371 is designed to be a peripherally restricted and selective CB2 receptor agonist, and is intended to provide pain relief without the unwanted side effects associated with CB1 receptor activation.
APD371 Development
We are conducting a Phase 2a clinical trial to evaluate APD371 in pain associated with CD. This exploratory study is an open‑label investigation to evaluate safety and tolerability of APD371 in this patient population and to gain initial insights into its efficacy via a pain visual analog scale, or VAS.
In April 2016, we announced favorable results from a Phase 1b multiple-ascending dose clinical trial of APD371. This randomized, double-blind, placebo-controlled Phase 1b clinical trial enrolled 36 healthy adults to evaluate the safety, tolerability and pharmacokinetics of multiple-ascending doses of APD371. Cohorts of 12 subjects (9 active, 3 placebo) were administered doses of 50 mg, 100 mg, or 200 mg of APD371 or placebo three times daily for 10 days and, in connection with the pharmacokinetic evaluation, one time on the 11th day. The most common adverse events were headache and nausea. All adverse events were classified as mild, and there were no serious adverse events reported. There was one discontinuation in the high-dose group due to an adverse event of mild thirst and somnolence. Reductions in blood pressure and heart rate were observed, but none were symptomatic or resulted in an adverse event. Drug levels at all doses tested in the trial, including the lowest dose, were well above those believed to be needed to stimulate the CB2 receptor.
In April 2015, we announced favorable top-line results from a Phase 1 single-ascending dose clinical trial of APD371. The randomized, double-blind and placebo-controlled trial enrolled 56 healthy adults to evaluate the safety, tolerability and pharmacokinetics of single-ascending doses of APD371. Dose-responsive exposure was observed over the explored dose range of 10-400 mg with good tolerability at all doses administered.
APD371 intellectual property
As of March 2, 2018, we owned issued patents covering compositions of matter for APD371 and related compounds in 21 jurisdictions, including the United States, China, Japan, Canada, Russia, South Korea and Australia, and we had applications pending in 10 other jurisdictions, of which the ones with the largest pharmaceutical markets were Europe, Venezuela, Brazil and India. The patent on APD371 issued by the US Patent and Trademark Office has serial number US 8,778,950. We also own issued patents and/or pending applications directed to various solid-state forms of APD371. The earliest priority date for the patents on APD371 is 2009. The terms of these patents are capable of continuing into 2030 in most jurisdictions without taking into account any patent term adjustment or extension regimes of any country or any additional term of exclusivity we might obtain by virtue of the later filed patent applications.
Additional Internal Preclinical and Clinical Programs
We have additional clinical and preclinical assets, including temanogrel and APD597, which we are evaluating for future development. We are also evaluating additional delivery forms of the products in our pipeline to extend clinical utility or improve the product profile.
Collaborations
In addition to our primary focus on internally developing our clinical pipeline, we have entered into strategic collaborations with pharmaceutical companies, including Everest Medicines Limited, or Everest, Axovant Sciences GmbH, or Axovant, Boehringer Ingelheim International GmbH, or Boehringer Ingelheim, Beacon Discovery, Inc., or Beacon, and Eisai.
Everest Collaboration
In December 2017, we entered into a Collaboration and License Agreement, or the Everest Agreement, with Everest.
Under the Everest Agreement, we granted Everest an exclusive, royalty-bearing license to develop, manufacture and commercialize two of our product candidates, ralinepag (in any formulation) and etrasimod (in oral formulations only), in China, Taiwan, Hong Kong, Macau and South Korea, or the Everest Territories.
Everest will be responsible for all development, manufacture and commercialization of the licensed products in the Everest Territories, and may participate in the portion of our global clinical trials that is conducted in the Everest Territories.
In addition to an upfront payment of $12.0 million, we are eligible to receive development, regulatory and commercial milestone payments from Everest of up to $212.0 million for both licensed products, as well as tiered royalties on net sales ranging from the high single digits to low double digits. Following an initial royalty term, we are eligible to receive a lower trademark royalty if Everest continues to use our licensed product-related trademarks.
Nelotanserin Collaboration
Nelotanserin, an orally available potent and selective inverse agonist of the 5-HT2A receptor, is an investigational drug candidate that has been implicated in the pathophysiology underlying psychosis. Nelotanserin was discovered by Arena, and we previously completed Phase 1 trials in healthy volunteers and Phase 2 trials in subjects with insomnia before development was discontinued for that indication.
Development, Marketing, and Supply Agreement
In May 2015, we entered into a Development, Marketing and Supply Agreement with Roivant Sciences Ltd., or Roivant, for nelotanserin. Roivant subsequently assigned all of its rights to develop and commercialize nelotanserin to its subsidiary, Axovant. Under our collaboration, Axovant has exclusive worldwide rights to develop and commercialize nelotanserin, and we are required to manufacture or have manufactured clinical supply and commercial product to sell to Axovant. We received a $4.0 million upfront payment and are eligible to receive $41.5 million in regulatory and development milestone payments. We are also eligible to receive 15% of net sales of nelotanserin in exchange for the manufacture and supply of finished commercial drug product, and up to a total of $60.0 million in one-time purchase price adjustment payments tied to certain commercial sales milestones.
Axovant has agreed to indemnify us for losses resulting from certain third-party claims, including for ![]() Axovant’s negligence, willful misconduct or violation of law, (ii) Axovant’s breach of the development, marketing and supply agreement or related agreements, (iii) any product liability claim, (iv) certain uses or misuses of nelotanserin, (v) certain infringement of intellectual property rights, and (vi) product manufactured according to the product warranty. We have agreed to indemnify Axovant for losses resulting from certain third-party claims, including for our negligence, willful misconduct or violation of law, and for losses resulting from our breach of the agreement or related agreements.
Axovant’s negligence, willful misconduct or violation of law, (ii) Axovant’s breach of the development, marketing and supply agreement or related agreements, (iii) any product liability claim, (iv) certain uses or misuses of nelotanserin, (v) certain infringement of intellectual property rights, and (vi) product manufactured according to the product warranty. We have agreed to indemnify Axovant for losses resulting from certain third-party claims, including for our negligence, willful misconduct or violation of law, and for losses resulting from our breach of the agreement or related agreements.
Axovant has the right to terminate the agreement on a compound-by-compound basis or in its entirety upon 90 days’ prior written notice to us. We have the right to terminate the agreement upon certain intellectual property concerns. Either party has the right to terminate the agreement early in certain circumstances, including if the other party is in material breach.
Nelotanserin development
Under our Development, Marketing and Supply Agreement, Axovant will be responsible for funding the development and commercialization of nelotanserin. In January 2018, Axovant reported results for a pilot Phase 2 Visual Hallucination study of nelotanserin in patients with Lewy body dementia. Additionally, Axovant is studying nelotanserin in patients with dementia with Lewy Bodies or Parkinson’s disease dementia who are experiencing rapid eye movement, or REM, sleep behavior disorder. Axovant will determine the overall development strategy for nelotanserin once its ongoing clinical, regulatory and commercial review is completed.
Orphan GPCR Collaboration
In December 2015, we entered into an exclusive agreement with Boehringer Ingelheim, to conduct joint research to identify drug candidates targeting a GPCR that belongs to a group of orphan central nervous system, or CNS, receptors. An “orphan receptor” is structurally related to a family of proteins that are known to act as functional cell-surface receptors but whose ligand has not yet been identified.
We contracted with Beacon to perform our research obligations under the Boehringer Ingelheim collaboration. In exchange, we agreed to share limited near term milestones with Beacon as well as the full-time equivalent funding paid to us by Boehringer Ingelheim. We have retained the longer-term success milestones and all royalties.
Beacon Discovery Agreements
In September 2016, we entered into a series of agreements with Beacon. Beacon was founded and is owned by several of our former employees.
We entered into a License and Collaboration Agreement with Beacon, pursuant to which we granted Beacon a non-exclusive, non-assignable and non-sublicensable license to certain database information relating to compounds, receptors and pharmacology, and transferred certain equipment to Beacon. Beacon will seek to engage global partners to facilitate discovery and development. Beacon has agreed to assign to us any intellectual property relating to our existing research and development programs developed in the course of performing research for us, and grant us a non-exclusive license to any intellectual property developed outside the course of performing work for us that is reasonably necessary or useful for developing or commercializing the products under our research and development programs. We are also entitled to rights of negotiation and rights of first refusal to potentially obtain licenses to certain compounds discovered and developed by Beacon. In addition, we are entitled to receive ![]() a percentage of any revenue received by Beacon on or after the second anniversary of the effective date of the agreement from any third party pursuant to a third-party license, including upfront payments, milestone payments and royalties; (ii) single-digit royalties on the aggregate net sales of any related products sold by Beacon and its affiliates; and (iii) in the event that Beacon is sold, a percentage of the consideration for such sale transaction.
a percentage of any revenue received by Beacon on or after the second anniversary of the effective date of the agreement from any third party pursuant to a third-party license, including upfront payments, milestone payments and royalties; (ii) single-digit royalties on the aggregate net sales of any related products sold by Beacon and its affiliates; and (iii) in the event that Beacon is sold, a percentage of the consideration for such sale transaction.
We also entered a Master Services Agreement with Beacon, pursuant to which Beacon performs certain research services for us relating to our proprietary pipeline, as well as a services agreement to support our research obligations under our collaboration with Boehringer Ingelheim.
BELVIQ (lorcaserin) Collaboration
Lorcaserin is approved for marketing in the United States, South Korea, Brazil, Mexico, Israel, and Taiwan for the indication of weight management, and is being commercialized by Eisai or its distributors in the United States, South Korea, Israel, and Taiwan. BELVIQ was made available by prescription in the United States in June 2013 and in South Korea in February 2015. Eisai also has launched of a once-daily formulation of lorcaserin in the United States, which is marketed under the brand name BELVIQ XR. Lorcaserin has not yet been launched in Brazil or Mexico. In December 2016, we entered into a Transaction Agreement and a Supply Agreement with Eisai, which replaced our prior marketing and supply agreement with Eisai for lorcaserin.
Transaction Agreement
Pursuant to the Transaction Agreement, we granted Eisai an exclusive, royalty-bearing license, or transferred intellectual property, to develop, manufacture and commercialize lorcaserin in all countries and territories of the world. In consideration for the rights granted to Eisai under the Transaction Agreement, Eisai has agreed to make tiered royalty payments to us on the net sales of lorcaserin. The royalty rates range from 9.5% on annual global net sales less than or equal to $175.0 million, 13.5% on annual global net sales greater than $175.0 million but less than or equal to $500.0 million and 18.5% on annual global net sales greater than $500.0 million.
We are eligible to receive a milestone payment of $25.0 million upon the achievement of global net sales of lorcaserin for a calendar year first exceeding $250.0 million.
Eisai is solely responsible for all costs and expenses in connection with the development of lorcaserin, and our wholly owned subsidiary, Arena Pharmaceuticals GmbH, or Arena GmbH, was relieved of its obligation under the former marketing and supply agreement to pay for its share of development costs for lorcaserin. Eisai has the exclusive right and responsibility to plan and implement all research and development of lorcaserin at its own cost and expense, including conducting all regulatory activities and all clinical and development activities. Additionally, Eisai has agreed to ![]() conduct all studies required by the FDA as a condition of obtaining and maintaining regulatory approval of lorcaserin in the United States (otherwise known as the cardiovascular outcomes trial, or CVOT), (ii) continue the current study assessing whether lorcaserin reduces the incidence of major cardiovascular events, (iii) continue the current study assessing whether lorcaserin reduces the incidence of conversion to Type 2 diabetes mellitus, and (iv) use commercially reasonable efforts to develop and seek regulatory approval of lorcaserin in each of China, Japan and the European Union.
conduct all studies required by the FDA as a condition of obtaining and maintaining regulatory approval of lorcaserin in the United States (otherwise known as the cardiovascular outcomes trial, or CVOT), (ii) continue the current study assessing whether lorcaserin reduces the incidence of major cardiovascular events, (iii) continue the current study assessing whether lorcaserin reduces the incidence of conversion to Type 2 diabetes mellitus, and (iv) use commercially reasonable efforts to develop and seek regulatory approval of lorcaserin in each of China, Japan and the European Union.
Eisai is solely responsible, and has the exclusive rights, for commercializing lorcaserin and is responsible for manufacturing lorcaserin, except for any manufacturing to be conducted by Arena GmbH under the Supply Agreement. Eisai is responsible for using commercially reasonable efforts to commercialize lorcaserin products in the United States, the European Union, China and Japan (collectively, the Major Markets) after regulatory approval in the applicable market.
We and Eisai will each bear 50% of losses arising from any alleged defective manufacturing of lorcaserin by Arena GmbH under the Supply Agreement, and Eisai will be solely responsible for any expenses and losses associated with other product liability claims.
The Transaction Agreement will remain in effect until terminated by us or Eisai with respect to all countries in the world. We may terminate the Transaction Agreement with respect to a Major Market if Eisai permanently ceases development and commercialization of lorcaserin products in such Major Market, or in its entirety if Eisai permanently ceases development and commercialization of lorcaserin products in the world. We may also terminate the Transaction Agreement if Eisai challenges any patent controlled by us related to lorcaserin as of the effective date of the Transaction Agreement, or Licensed Patents, if Eisai is debarred under the US Federal Food, Drug, and Cosmetic Act, or if Eisai is in material breach of the standstill provisions. Eisai may terminate the Transaction Agreement if as a result of its change of control, it would be in breach of certain competition restrictions.
In the event the Transaction Agreement is terminated by us due to Eisai’s failure to develop and commercialize lorcaserin products, Eisai’s challenging of any of the Licensed Patents or Eisai’s debarment or material breach of the standstill provisions, or by Eisai after a change of control that would result in Eisai being in breach of certain competition restrictions, Eisai will grant Arena an exclusive, royalty-free license to certain patent rights and know-how necessary or useful for the development and commercialization of lorcaserin products, re-assign the assets purchased by Eisai under the Transaction Agreement and Supply Agreement, and provide certain other transition assistance.
Supply Agreement
Under the Supply Agreement, Arena GmbH agreed to manufacture and supply, and Eisai agreed to purchase, all of Eisai’s requirements (or specified minimum quantities if such quantities are greater than Eisai’s requirements), subject to certain exceptions, for BELVIQ and BELVIQ XR for the development and commercial use of such products in all countries and territories of the world for an initial two-year period, which initial period may be extended by Eisai for an additional payment. Eisai will pay Arena GmbH agreed upon prices to deliver finished drug product during this time. Additionally, Eisai agreed to pay up to CHF 13.0 million in payments to Arena GmbH to support the maintenance of Arena GmbH’s manufacturing facility in Switzerland during the initial two-year supply period, and an additional amount during the extension period, if any.
Pursuant to the Supply Agreement, Arena GmbH agreed to transfer to Eisai all know-how and materials necessary for Eisai to manufacture BELVIQ at the facility in accordance with Arena GmbH’s manufacturing processes used at the effective date of the Supply Agreement or 24 months prior. Arena GmbH also assigned its agreements with distributors in South Korea, Taiwan and Israel to Eisai, and Eisai agreed to assume responsibilities under such agreements. On the effective date of the Supply Agreement, Eisai purchased Arena GmbH’s entire inventory of the precursor materials for manufacturing lorcaserin then in Arena GmbH’s possession. In exchange for these materials Eisai made a one-time payment to Arena GmbH of $10.0 million.
Absent early termination, the Supply Agreement will remain in effect until ![]() the last day of the initial two-year supply period, or the last day of the six-month extension period (if any), or up to two weeks thereafter if so requested by Eisai, or (ii) in the event of an acquisition of Arena or Arena GmbH by a third party, or of an assignment of the Supply Agreement by Arena GmbH to a third party, five years after the effective date of the Supply Agreement. After the initial two-year period of the Supply Agreement, either Arena GmbH or Eisai may terminate the Supply Agreement upon the other party’s material breach that remains uncured 60 days after receiving written notice thereof. The Supply Agreement will also terminate automatically upon termination of the Transaction Agreement.
the last day of the initial two-year supply period, or the last day of the six-month extension period (if any), or up to two weeks thereafter if so requested by Eisai, or (ii) in the event of an acquisition of Arena or Arena GmbH by a third party, or of an assignment of the Supply Agreement by Arena GmbH to a third party, five years after the effective date of the Supply Agreement. After the initial two-year period of the Supply Agreement, either Arena GmbH or Eisai may terminate the Supply Agreement upon the other party’s material breach that remains uncured 60 days after receiving written notice thereof. The Supply Agreement will also terminate automatically upon termination of the Transaction Agreement.
On March 9, 2018, we entered into an Asset Purchase Agreement, or Sale Agreement, with Siegfried Pharma AG and Siegfried AG (collectively and individually, Siegfried). Under the Sale Agreement, we agreed to sell and assign to Siegfried, and Siegfried agreed to purchase and assume from Arena GmbH, certain drug product finishing facility assets and know-how, including fixtures, equipment, other personal property and real estate assets located in Zofingen, Switzerland, related contracts and certain related liabilities after the closing as well as to the transfer of all of Arena GmbH’s approximately 50 current employees, or collectively, Manufacturing Operations. We refer to this transaction as Siegfried Transaction. Under the Sale Agreement, at the closing, Arena GmbH will assign to Siegfried Pharma the Supply Agreement.
Intellectual Property
Our success depends in large part on our ability to protect our compounds and information, and to operate without infringing the proprietary rights of third parties. We rely on a combination of patent, trade secret, copyright, and trademark laws, as well as confidentiality, licensing and other agreements, to establish and protect our proprietary rights. We seek patent protection for our key inventions, including drug candidates we identify, routes for chemical synthesis, pharmaceutical formulations and methods of treatment.
There is no assurance that any of our patent applications will issue, or that any of the patents will be enforceable or will cover a drug or other commercially significant product or method. In addition, we regularly review our patent portfolio to identify patents and patent applications for potential abandonment that we deem to have relatively low value to our ongoing business operations. There is also no assurance that we will correctly identify which of our patents and patent applications should be maintained and which should be abandoned. The term of most of our other current patents commenced, and most of our future patents, if any, will commence, on the date of issuance and terminate 20 years from the earliest effective filing date of the patent application. Because any marketing and regulatory approval for a drug often occurs several years after the related patent application is filed, the resulting market exclusivity afforded by any patent on our drug candidates will likely be substantially less than 20 years.
In the United States, patent term adjustment is available for certain delays in patent office proceedings. In addition, under the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, the term of a patent that covers an FDA-approved drug may be eligible for patent term extension, or PTE. PTE permits patent term restoration of a US patent as compensation for the patent term lost during product development and the FDA regulatory review process. The Hatch-Waxman Act permits a PTE of up to five years beyond the expiration of the patent. This period is generally one-half the time between the effective date of an Investigational New Drug, or IND (falling after issuance of the patent), and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application, provided the sponsor acted with diligence. The Improving Regulatory Transparency for New Medical Therapies Act was signed into law in 2015 to prevent the loss of PTE (and market exclusivity) for drugs for which the FDA recommends scheduling under the Controlled Substances Act. A PTE cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. The application for PTE is subject to approval by the PTO in conjunction with the FDA.
Outside of the United States, similar provisions may be available in the European Union, Japan, South Korea and some other jurisdictions to extend the term of a patent that covers an approved drug. The length of any such extension would vary by country. Our European patents may be eligible for supplemental protection certificates of up to five years in one or more countries.
Due to the specific requirements for obtaining these extensions, there is no assurance that our patents will be afforded extensions even if we encounter significant delays in patent office proceedings or marketing and regulatory approval.
In addition to patent protection, we rely on trade secrets, proprietary know-how and continuing technological advances to develop and maintain our competitive position. To maintain the confidentiality of our trade secrets and proprietary information, all of our employees are required to enter into and adhere to an employee confidentiality and invention assignment agreement, and invention disclosure procedures as a condition of employment. Additionally, our employee confidentiality and invention assignment agreements require that our employees not bring to us, or use without proper authorization, any third-party proprietary technology. We also generally require our consultants and collaborators that have access to proprietary property and information to execute confidentiality and invention rights agreements in our favor before beginning their relationship with us. While such arrangements are intended to enable us to better control the use and disclosure of our proprietary property and provide for our ownership of proprietary technology developed on our behalf, they may not provide us with meaningful protection for such property and technology in the event of unauthorized use or disclosure.
Competition
The biotechnology and pharmaceutical industries are highly competitive and are subject to rapid and significant change. We face significant competition from many organizations with drugs or drug candidates that do or may compete drug candidates we are developing. We may not be able to compete successfully against these organizations, which include many large, well-financed and experienced pharmaceutical and biotechnology companies, as well as academic and research institutions and government agencies. Developments by others may render our drug candidates obsolete or noncompetitive, and we or our collaborators may not be successful in developing either first or best in class drugs.
Many of our existing and potential competitors have substantially greater drug development capabilities and financial, scientific and marketing resources than we do. Additional consolidation in the pharmaceutical industry may result in even more resources being concentrated with our competitors. As a result, our competitors may be able to devote greater resources than we can to the research, development, marketing and promotion of therapeutic products or drug discovery techniques, or to adapt more readily to technological advances than we can. Accordingly, our competitors may succeed in obtaining patent protection, receiving regulatory approval or commercializing drugs before we do.
We expect to encounter significant competition in the therapeutic areas targeted by our principal drug candidates. Companies that complete clinical trials, obtain regulatory approvals and commence commercial sales of their drug candidates before us may achieve a significant competitive advantage. Furthermore, we may be competing against companies with substantially greater manufacturing, marketing, distribution and selling capabilities, and any drug candidate that we successfully develop may compete with existing therapies that have longer histories of safe and effective use.
We may rely on collaborators for support of development programs and for the manufacturing and marketing of drug candidates. Such collaborators may be conducting multiple drug development efforts within the same disease areas that are the subject of their agreements with us, which may negatively impact the development of drugs that are subject to our agreements. In addition, we face and will continue to face intense competition from other companies for such collaboration arrangements, and technological and other developments by others may make it more difficult for us to establish such relationships.
Research and Development Expenses
Research and development activities are the primary source of our expenses. Our research and development expenses include personnel costs, facility and equipment costs, clinical and preclinical study fees, research supplies, and manufacturing costs for non-commercial products. Such expenses totaled $71.0 million, $63.8 million, and $83.2 million for the years ended December 31, 2017, 2016, and 2015, respectively. For research and development sponsored by collaborators for which we initially incur the costs, we record the costs within research and development expenses and record the reimbursements we receive from the collaborators for these costs within revenues; these expenses and revenues totaled $2.9 million, $2.8 million, and $1.2 million for the years ended December 31, 2017, 2016, and 2015, respectively.
Employees
As of March 9, 2018, we had a total of 159 employees, including 122 in research, development and manufacturing and 37 in administration, which includes finance, legal, facilities, information technology and other general support areas. This includes the approximately 50 employees that we expect will be transferred to Siegfried as part of the sale of our manufacturing operations in Switzerland.




