Clovis Oncology
Overview
Clovis Oncology (CLVS) is a biopharmaceutical company focused on acquiring, developing and commercializing innovative anti-cancer agents in the United States, Europe and additional international markets. The company target its development programs for the treatment of specific subsets of cancer populations, and simultaneously develop, with partners, diagnostic tools intended to direct a compound in development to the population that is most likely to benefit from its use.
The company's marketed product Rubraca® (rucaparib) is approved on an accelerated basis in the United States by the Food and Drug Administration (“FDA”) as monotherapy for the treatment of patients with deleterious BRCA (human genes associated with the repair of damaged DNA) mutation (germline and/or somatic) associated advanced ovarian cancer who have been treated with two or more chemotherapies, and selected for therapy based on an FDA-approved companion diagnostic for Rubraca. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. The company launched Rubraca in the United States in December 2016 for this indication.
The FDA is currently reviewing on a priority review timeline its supplemental New Drug Application (“sNDA”) for Rubraca as maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy.
The company's Marketing Authorization Application (“MAA”) submitted to the European Union’s European Medicines Agency (“EMA”) for an ovarian cancer treatment indication for Rubraca is currently under review by the EMA’s Committee for Medicinal Products for Human Use (“CHMP”). Following a Scientific Advisory Group - Oncology meeting and an oral explanation by it before the CHMP in February 2018, the CHMP has communicated a positive trend vote for the MAA and their intention to hold a final vote on the treatment indication at their March 2018 meeting.
Beyond its labeled indication, Clovis Oncology has a robust Rubraca clinical development program underway in a variety of solid tumor types, also including prostate and bladder cancers, and in July 2017, the company entered into a broad clinical collaboration with Bristol-Myers Squibb Company to evaluate the combination of their immunotherapy Opdivo® (nivolumab) with Rubraca in several tumor types. The company hold worldwide rights for Rubraca.
In addition, Clovis Oncology has two other product candidates: lucitanib, an oral inhibitor of the tyrosine kinase activity of vascular endothelial growth factor receptors (“VEGFR”) 1-3, platelet-derived growth factor receptors (“PDGFR”) alpha and beta and fibroblast growth factor receptors (“FGFR”) 1-3, and rociletinib, an oral mutant-selective inhibitor of epidermal growth factor receptor (“EGFR”). While Clovis Oncology has stopped enrollment in ongoing trials for each of these candidates, the company continue to provide drug to patients whose clinicians recommend continuing therapy. The company maintain certain development and commercialization rights for lucitanib and global development and commercialization rights for rociletinib.
Clovis was founded in 2009. Clovis Oncology has built its organization to support innovative oncology drug development for the treatment of specific subsets of cancer populations. To implement its strategy, Clovis Oncology has assembled an experienced team with core competencies in global clinical and non-clinical development, regulatory operations and commercialization in oncology, as well as conducting collaborative relationships with companies specializing in companion diagnostic development.
Rubraca – a PARP Inhibitor
Overview
Rubraca (rucaparib) is an oral, small molecule poly ADP-ribose polymerase (“PARP”) inhibitor of PARP1, PARP2 and PARP3. The company in-licensed Rubraca from Pfizer Inc. in June 2011. In the United States, Rubraca is approved by the FDA for the treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer who have been treated with two or more chemotherapies, and selected for therapy by an FDA-approved companion diagnostic for Rubraca. The indication is approved under the FDA’s accelerated approval program based on objective response rate and duration of response, and is based on results from two multicenter, single-arm, open-label clinical trials. Continued approval for this indication may be contingent upon verification and description of clinical benefit in ARIEL3 and/or ARIEL4, its confirmatory trials. Foundation Medicine, Inc. has developed two companion diagnostics that are FDA-approved and commercially available for selection of patients for treatment with Rubraca in this indication: FoundationFocus™ CDxBRCA and FoundationOne CDx™.
In October 2017 the company submitted a sNDA for Rubraca for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy. In December 2017, the FDA accepted its sNDA for Rubraca and granted priority review status to the application with a Prescription Drug User Free Act (“PDUFA”) goal date of April 6, 2018. If approved, the company expect that diagnostic testing would not be required for patients to be prescribed Rubraca in this maintenance treatment indication. Clovis Oncology has approximately 150 field-based personnel in the United States, and these commercial and medical affairs organizations already in place will support the commercial launch of Rubraca, if approved in this expanded maintenance treatment indication.
The second-line maintenance treatment paradigm in ovarian cancer is being rapidly adopted in the U.S. following the approval of two other PARP inhibitors in this setting during 2017. If Rubraca is approved in this setting, with no requirement for diagnostic testing as supported by the ARIEL3 data, it would be possible to address a patient population approximately four times larger than its initial niche treatment indication, which is limited to women with ovarian cancer in the third-line setting with a BRCA mutation. BRCA mutations are believed to occur in approximately 25 percent of women with ovarian cancer.
The company's MAA submitted to the EMA for an ovarian cancer treatment indication is currently under review. Following a Scientific Advisory Group - Oncology meeting and an oral explanation before the CHMP in February 2018, the CHMP communicated to it that they held a trend vote for the MAA and the result was positive. The CHMP also communicated to it their intention to hold a final vote on the treatment indication at their March 2018 meeting. The CHMP’s trend vote is not binding on the CHMP with respect to the final vote. The indication currently under consideration by the CHMP focuses on a subset of platinum-sensitive ovarian cancer where there is particular unmet need. If Rubraca receives a favorable opinion from the CHMP in March, a potential approval from the European Commission is expected during the second quarter of 2018. In the event of such an approval for the treatment indication, the company intend to submit a variation to the marketing authorization (“MA”) for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy, for which the company anticipate a CHMP opinion may come during the fourth quarter of 2018. The EMA’s Committee for Orphan Medicinal Products (“COMP”) will meet in March 2018 to review and issue a final opinion on its application to maintain the orphan drug designation for Rubraca submitted as part of the MAA review process. While Rubraca currently has orphan drug designation in the EU, there is no assurance that COMP will maintain such designation post-approval. In the event of a negative opinion for the treatment indication, the company expect to file a new MAA for the maintenance treatment indication during the second quarter of 2018. If approved in the EU, the company intend to commercialize Rubraca on its own and Clovis Oncology is building its commercial and medical affairs infrastructure in Europe. The leadership of those teams is in place, and the company intend to hire field sales personnel as pricing and reimbursement decisions are made and approvals are received.
The Role of PARP Inhibition in Cancer Therapy
Cells in the human body are under constant attack from agents that can cause damage to DNA, including sunlight and other forms of radiation, as well as DNA-binding chemicals that can cause changes in the composition of DNA. Cells have evolved multiple mechanisms to enable such DNA repair, and these mechanisms are complementary to each other, each driving repair of specific types of DNA damage. If a cell’s DNA damage repair system is overwhelmed, then the cell will die undergoing a form of suicide termed apoptosis. A fundamental principle of cancer therapy is to damage cells profoundly with radiation or DNA-binding drugs, such as alkylating agents or platinums, to induce apoptosis and, subsequently, cancer cell death. Multiple DNA repair mechanisms active in the cell may reduce the activity of these anti-cancer therapies.
The PARP family comprises 17 structurally related proteins that have been identified on the basis of sequence similarity. PARP1, PARP2, and PARP3 play a central role in DNA repair. They are rapidly recruited to the sites of DNA damage and catalyze the recruitment of additional proteins that initiate the repair of damaged DNA. The breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes also have important roles in DNA repair pathways such as homologous recombination. According to the National Cancer Institute, BRCA1 and BRCA2 mutations are associated with an increased risk of ovarian, breast, prostate, and pancreatic cancers.
Rubraca is an inhibitor of PARP enzymes, including PARP1, PARP2, and PARP3. PARP inhibitors have shown activity in BRCA1/2 mutant and homologous recombination (“HR”) repair deficient cancer cell lines through a mechanism known as synthetic lethality in which the loss of two genes/pathways is required for cell death. The inhibition/inactivation of repair pathways by administration of a PARP inhibitor in the context of an underlying genetic defect such as a BRCA mutation results in tumor cell death through accumulation of unrepaired DNA damage.
Alterations in DNA repair genes other than BRCA1/2 have been observed in, and contribute to the hereditary risk of, ovarian, breast, prostate and pancreatic cancers. PARP inhibitors have shown evidence of nonclinical and clinical activity in tumors with alterations in non-BRCA HR genes. DNA repair deficiencies resulting from genetic and epigenetic alterations can result in a “BRCA-like” phenotype that may also render tumor cells sensitive to PARP inhibitors. One approach to identify patients with DNA repair deficiencies due to mechanisms other than a mutation in BRCA or other non-BRCA HR genes is to assess loss of heterozygosity (“LOH”), or the loss of one normal copy of a gene, which arises from error-prone DNA repair pathways when HR is compromised.
On the basis of these scientific observations, the company initially developed Rubraca in ovarian cancer patients with tumors having BRCA mutations or other homologous recombination deficiencies (“HRD”). These molecular markers also may be used to select patients with other tumors for treatment with Rubraca. Thus, in addition to ovarian trials, studies open for enrollment or under consideration to further evaluate Rubraca, either alone or in combination with other agents, include prostate, breast, pancreatic, bladder, gastroesophageal and lung cancers.
Rubraca Clinical Development
Clovis Oncology is developing Rubraca for selected patient populations and collaborating with partners for companion diagnostic development. Clovis Oncology has focused its development strategy for Rubraca on indications where the company believe patient populations exhibit higher frequencies of mutant BRCA tumors or HRD, where PARP inhibitors have demonstrated clinical or pre-clinical activity in tumors. In certain of these trials, the company or its partners will have access to interim data on a periodic or continuing basis that will not be made available publicly on the same timeframe as such data becomes available to it, or at all. The following table summarizes the ongoing Clovis- or partner-sponsored studies:
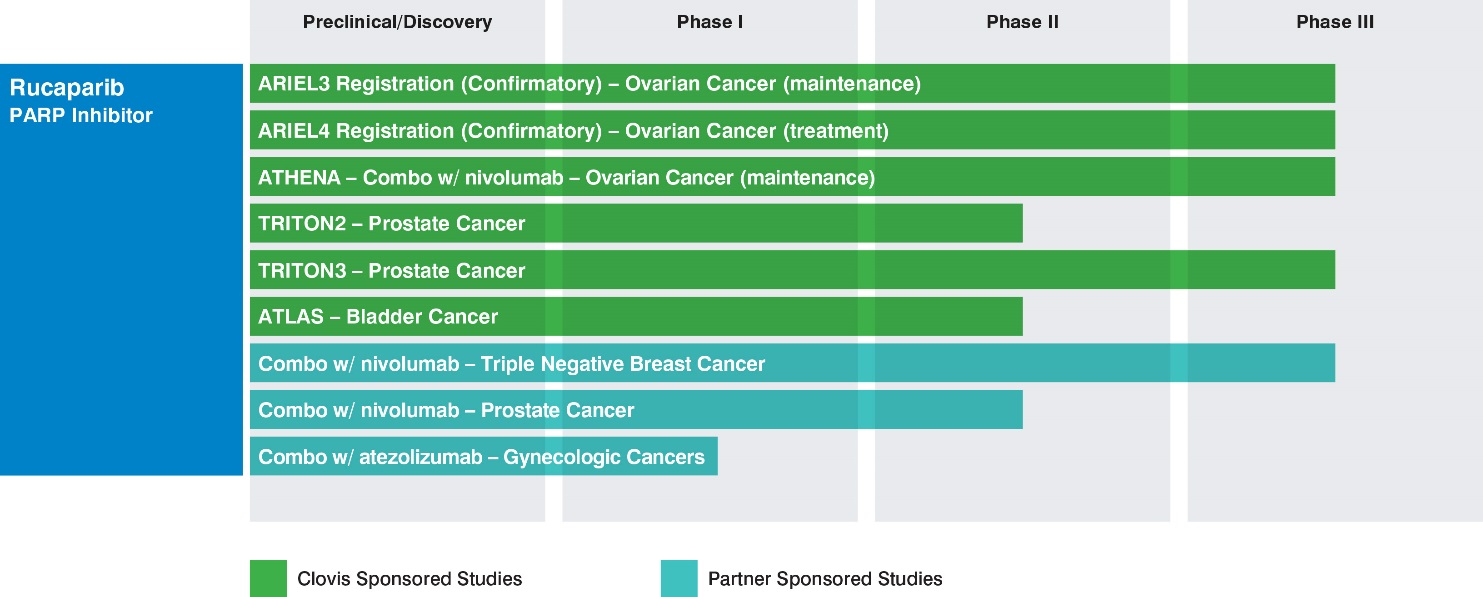
Ovarian cancer
According to the American Cancer Society, an estimated more than 22,000 women were diagnosed with ovarian cancer in the United States in 2017, and according to GLOBOCAN in 2012, an estimated more than 65,000 women in Europe are diagnosed each year with ovarian cancer, and ovarian cancer is among those cancers with the highest rate of deaths. Approximately 80% to 85% of ovarian cancer cases are not diagnosed, and therefore remain untreated, until the disease has spread to other parts of the body, or metastasized. Most women with ovarian cancer will relapse after surgery and/or chemotherapy. BRCA mutations, either germline or somatic, are believed to occur in approximately 25 percent of women with ovarian cancer according to an article published in Clinical Cancer Research in 2014.
The ARIEL (Assessment of Rucaparib In Ovarian CancEr TriaL) program is a novel, integrated translational-clinical program designed to accurately and prospectively identify ovarian cancer patients with tumor genotypes associated with benefit from Rubraca therapy.
ARIEL2 (NCT01891344) is a two-part single-arm open label study designed to identify pre-specified tumor characteristics that predict sensitivity to Rubraca using DNA sequencing to evaluate each patient’s tumor. Part 1 enrolled 204 platinum-sensitive patients and updated results were presented in June 2016. Part 2 enrolled 286 patients with advanced ovarian cancer who have received at least three prior chemotherapy regimens and includes platinum-sensitive, -resistant and -refractory patients. ARIEL2 is evaluating clinical response in patients classified into molecularly-defined subgroups, including germline BRCA-mutant, somatic BRCA-mutant and HRD by a prospectively defined genomic signature.
The efficacy of Rubraca in the ovarian cancer treatment setting was assessed in 106 patients from ARIEL2 and Study 10 (NCT01482715, a multicenter, single-arm, open-label clinical trial of Rubraca), in patients with advanced BRCA-mutant ovarian cancer who had progressed after two or more prior chemotherapies. Median age was 59 years and median number of prior chemotherapy regimens was three. All 106 patients received the starting dose of Rubraca 600 mg twice daily. The major efficacy outcome measure of both trials was objective response rate (ORR) and duration of response (DOR) as assessed by the investigator according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. All responses were confirmed. ORR assessed by investigator was 54% (95% Confidence Interval, or CI: 44, 64), with a median DOR of 9.2 months (95% CI: 6.6, 11.6). ORR by independent radiology review was 42% (95%: 32, 52), with a median DOR of 6.7 months (95% CI: 5.5, 11.1).
The overall safety evaluation of Rubraca in the advanced ovarian cancer treatment setting is based on data from 377 patients with ovarian cancer from ARIEL2 and Study 10. The most common adverse reactions (≥ 20% of patients; Grade 1-4) were nausea, asthenia/fatigue, vomiting, anemia, constipation, dysgeusia, decreased appetite, diarrhea, abdominal pain, thrombocytopenia and dyspnea. The most common laboratory abnormalities (≥ 35% of patients; Grade 1-4) were increase in creatinine, increase in aspartate aminotransferase (“AST”) levels, increase in alanine aminotransferase (“ALT”) levels, decrease in hemoglobin, decrease in lymphocytes, increase in cholesterol, decrease in platelets and decrease in absolute neutrophil count. Elevations in ALT/AST concentrations were generally self-limiting and not associated with other signs of liver toxicity. Rubraca requires monitoring of complete blood counts at baseline, and monthly thereafter. The most common Grade 3-4 adverse reaction was anemia, and the most common Grade 3-4 laboratory abnormality was a decrease in hemoglobin. Myelodysplastic Syndrome/Acute Myeloid Leukemia, or MDS/AML, was reported in two of the 377 (0.5%) patients with ovarian cancer treated with Rubraca. Both of these patients had received prior treatment with platinum and other DNA damaging agents. In addition, AML was reported in two (<1%) patients with ovarian cancer enrolled in ARIEL3. One case of AML was fatal. Both patients had received prior treatment with platinum and other DNA damaging agents.
The data from the two studies formed the basis of its submission of a New Drug Application (“NDA”) in the United States to the FDA in late June 2016. The application was granted priority review and approved under FDA’s accelerated approval program on December 19, 2016 as monotherapy for the treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer, who have been treated with two or more chemotherapies, and selected for therapy based on an FDA-approved companion diagnostic for Rubraca. Continued approval for this indication may be contingent upon verification and description of clinical benefit in ARIEL3 and/or ARIEL4, its confirmatory trials.
The efficacy of Rubraca in the ovarian cancer maintenance treatment setting was investigated in ARIEL3 (NCT01968213), a double-blind, multicenter clinical trial in which 564 patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who were in response to platinum-based chemotherapy were randomized (2:1) to receive Rubraca tablets 600 mg orally twice daily (n=375) or placebo (n=189). Treatment was continued until disease progression or unacceptable toxicity. All patients had achieved a response (complete or partial) to their most recent platinum-based chemotherapy. Randomization was stratified by best response to last platinum (complete or partial), time to progression following the penultimate platinum therapy (6 to < 12 months and ≥ 12 months), and tumor biomarker status. The major efficacy outcome was investigator-assessed progression-free survival (“PFS”) evaluated according to RECISTv1.1.
The primary efficacy analysis evaluated three prospectively defined molecular sub-groups in a step-down manner: 1) tumor BRCA mutant (tBRCAmut) patients, inclusive of germline and somatic BRCA mutations (n=196); 2) HRD patients, including tBRCAmut patients and BRCA wild-type with high LOH (n=354), and, finally, 3) the intent-to-treat population, or all patients treated in ARIEL3 (n=564). ARIEL3 demonstrated a statistically significant improvement in PFS for patients randomized to Rubraca as compared with placebo in all patients, and in the HRD and tBRCAmut subgroups. Median PFS in the tBRCAmut patients was 16.6 months (95% CI: 13.4–22.9) in the Rubraca group (n=130) versus 5.4 months (95% CI: 3.4–6.7) in the placebo group (n=66) (Hazard Ratio, or HR: 0.23 [95% CI: 0.16–0.34]; p<0.0001). Median PFS in the HRD patients was 13.6 months (95% CI: 10.9–16.2) in the Rubraca group (n=236) versus 5.4 months (95% CI: 5.1–5.6) in the placebo group (n=118) (HR: 0.32 [95% CI: 0.24–0.42]; p<0·0001). Median PFS in the intent-to-treat population was 10.8 months (95% CI: 8.3–11.4) in the Rubraca group (n=375) versus 5.4 months (95% CI: 5.3–5.5) in the placebo group (n=189) (HR: 0.36 [95% CI: 0.30–0.45]; p<0·0001).
Results from a blinded independent radiology review (“BICR”) were consistent. In a pre-specified analysis of the key stand-alone secondary endpoint of progression-free survival assessed by BICR, PFS was also improved in the Rubraca group compared with placebo in all three populations. Median PFS in the tBRCAmut patients was 26.8 months (95% CI: 19.2 to not reached) in the Rubraca group versus 5.4 months (95% CI: 4.9–8.1) in the placebo group (HR: 0.20 [95% CI: 0.13–0.32]; p<0.0001). Median PFS in the HRD patients was 22.9 months (95% CI: 16.2 to not reported) in the Rubraca group versus 5.5 months (95% CI: 5.1–7.4) in the placebo group (HR: 0.34 [95% CI: 0.24–0.47]; p<0.0001). Median PFS in the intent-to-treat population was 13.7 months (95% CI: 11.0–19.1) versus 5.4 months (95% CI: 5.1–5.5) in the placebo group (HR: 0.35 [0.28–0.45]; p<0.0001).
Enrollment in ARIEL3 included one-third of patients who had achieved a complete response to their prior platinum-based therapy, and two-thirds of patients who had achieved a partial response to their prior platinum-based therapy. Of those with a partial response, 37% had measurable disease at the time of enrollment and were therefore evaluable for response. The confirmed overall response rate by investigator-assessed RECISTv1.1 in the tBRCAmut group treated with Rubraca was 37.5% (15/40), of these, 17.5% (7/40) were complete responses. This compared with 9% (2/23) in the placebo group (p=0.0055). No complete responses were seen in the tBRCAmut placebo group. RECIST responses were also observed in BRCA wild-type HRD-positive and BRCA wild-type HRD-negative subgroups. RECIST responses were not assessed by independent blinded review.
Safety data from ARIEL3 demonstrated consistency with prior Rubraca studies. Treatment emergent adverse events (“TEAEs”) in the ARIEL3 Rubraca group were generally managed with dose modifications and not associated with increased mortality or morbidity compared with the placebo group. The most common (occurring in ≥5% of patients) TEAEs of grade ≥3 reported in patients treated with Rubraca in the ARIEL3 study were anemia/decreased hemoglobin (21%), increase in ALT/AST (10%), neutropenia (7%), asthenia/fatigue (7%) and thrombocytopenia (5%). The discontinuation rate for TEAEs (excluding disease progression) was 15% for Rubraca-treated patients and 2% for the placebo arm. In ARIEL3, the rate of treatment-emergent myelodysplastic syndrome (“MDS”)/acute myeloid leukemia (“AML”) in the Rubraca arm was <1% (3/372), and no patients on the placebo arm experienced treatment-emergent MDS/AML. In approximately 1,100 patients treated with Rubraca, MDS/AML occurred in 10 patients (0.9%), including those in long term follow-up. Of these, 5 occurred during treatment or during the 28 day safety follow-up (0.5%). The duration of Rubraca treatment prior to the diagnosis of MDS/AML ranged from 1 month to approximately 28 months. The cases were typical of secondary MDS/cancer therapy-related AML; in all cases, patients had received previous platinum-containing chemotherapy regimens and/or other DNA damaging agents.
At the time of the analysis of PFS, overall survival (OS) data were not mature (with 22% of events). The comprehensive dataset for ARIEL3 was presented at the 2017 European Society of Medical Oncology (“ESMO”) Congress in early September 2017 and subsequently published in The Lancet.
Based on the ARIEL3 dataset, in October 2017 the company submitted a supplemental NDA for Rubraca as maintenance treatment in adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy. The application was granted priority review status by the FDA and has a PDUFA date of April 6, 2018.
The ARIEL4 confirmatory study (NCT 02855944), which is open for enrollment, is a Phase 3 multicenter, randomized study of Rubraca versus chemotherapy in relapsed ovarian cancer patients with BRCA mutations (inclusive of germline and/or somatic) who have failed two prior lines of therapy. The primary endpoint of the study is PFS.
The Phase 1 RUCA-J study, sponsored by it, recently initiated with the first patient dosed with Rubraca in Japan. The Phase 1 study seeks to identify the recommended dose of rucaparib in Japanese patients.
Prostate cancer
The American Cancer Society estimates that more than 164,000 men in the United States will be diagnosed with prostate cancer in 2018, and the GLOBOCAN Cancer Fact Sheets estimated that approximately 345,000 men in Europe were diagnosed with prostate cancer in 2012. Castrate-resistant prostate cancer has a high likelihood of developing metastases. Metastatic castrate-resistant prostate cancer, or mCRPC, is an incurable disease, usually associated with poor prognosis. According to the American Cancer Society, the five-year survival rate for mCRPC is approximately 29%. Germline or somatic mutations in BRCA, ATM and other DNA repair genes are believed to be present at frequencies of 20 percent or higher in mCRPC, according to an article published in JCO Precision Oncology in 2017. These molecular markers may be used to select patients for treatment with a PARP inhibitor. Additionally, preclinical studies of rucaparib have demonstrated activity in prostate cell cancer lines deficient in BRCA or ATM.
The TRITON (Trial of Rucaparib in Prostate Indications) program in prostate cancer initiated in the second half of 2016, and currently includes two Clovis-sponsored potential registration studies currently open for enrollment:
The TRITON2 study (NCT02952534), a Phase 2 single-arm study of Rubraca in men with mCRPC enrolling patients with BRCA mutations and ataxia-telangiectasia mutations, or ATM, (both inclusive of germline and/or somatic) or other deleterious mutations in other homologous recombination repair genes. All patients will have progressed after receiving one line of taxane-based chemotherapy and one or two lines of androgen receptor (“AR”) targeted therapy in the castrate-resistant setting. The primary endpoints of the study are radiologic ORR in patients with measurable disease and protein-specific antigen (“PSA”) response rate in patients who do not have measurable disease. TRITON2 initiated during the fourth quarter of 2016, and interim data are expected at a medical meeting during the second half of 2018. The estimated primary completion date for TRITON2 is the second half of 2019. Pending positive data, TRITON2 could potentially serve as the basis for an accelerated approval in the U.S.The TRITON3 study (NCT02975934), a Phase 3 comparative study in men with mCRPC enrolling BRCA mutant and ATM (both inclusive of germline and/or somatic) patients who have progressed on AR-targeted therapy and who have not yet received chemotherapy in the castrate-resistant setting. TRITON3 will compare Rubraca to physician’s choice of AR-targeted therapy or chemotherapy in these patients. The planned primary endpoint of the study is radiologic PFS. TRITON3 initiated during the first quarter of 2017, and this earlier-line comparative study could potentially serve as a confirmatory study in the advanced prostate setting.
Bladder cancer
According to GLOBOCAN Cancer Fact Sheets, bladder cancer was one of the top six most common cancers in the United States as of 2012, with an estimated 79,000 new cases of bladder cancer diagnosed in the United States in 2017, according to the National Cancer Institute. Approximately 20% to 30% of newly diagnosed bladder cancer patients have disease that has invaded the muscle, according to the National Cancer Institute. Muscle-invasive bladder cancer is a disease with poor prognosis. Overall survival of these patients after initial cisplatin-containing chemotherapy is 13-15 months, with most patients experiencing relapse of disease in 9 months, according to an article published in the European Journal of Cancer in 2006. After the first one or two lines of anti-cancer treatments, options for these patients are limited, with platinum therapy as the current standard of care. Based an analysis of The Cancer Genome Atlas (“TCGA”), bladder cancer data set, the company believe approximately 60% of bladder cancer tumors have alterations in homologous recombination repair genes or other genomic features associated with HRD.
The company initiated a potential registration study in bladder cancer during the first quarter of 2018, called ATLAS (A Study of Rucaparib in Patients with Locally Advanced or Metastatic Urothelial Carcinoma). ATLAS (NCT03397394) is a phase 2 single-arm study enrolling patients with relapsed metastatic urothelial carcinoma following one or two prior standard of care regimens, with measurable disease, and no prior PARP treatment. The planned primary endpoint is overall response rate, and the study will enroll all comers, with no selection based on HRD status. Pending positive data, the company believe this trial design could support a sNDA in an all comers population, without regard to biomarker status.
Combination trials
In July 2017, the company and Bristol-Myers Squibb Company (“BMS”) announced a broad clinical collaboration evaluating the combination of Rubraca with BMS’s immunotherapy Opdivo® (nivolumab) in multiple tumor types. Three trials are underway or expected to initiate in the first half of 2018:
The ATHENA study, a phase 3 study in advanced ovarian cancer, sponsored by Clovis;A phase 3 study in advanced triple-negative breast cancer (TNBC), sponsored by BMS; andA phase 2 study in mCRPC sponsored by BMS.
The company believe that a preclinical rationale supports the conduct of clinical trials of the combination of its PARP inhibitor Rubraca with immune checkpoint inhibitors such as the PD-1 inhibitor Opdivo. BRCA1 and BRCA2 and other HRD mutations are associated with increased tumor mutational burden, which may create additional tumor-specific antigens or “neoepitopes.” Increased tumor mutation burden has been shown to correlate with increased benefit from immune checkpoint blockade. In addition, cell death that is induced by a PARP inhibitor is considered immunogenic, and stimulates a “STING-like” pathway due to fragmented DNA release into cytosol. In mice studies, rucaparib and an anti- PD-1 antibody demonstrated anti-tumor activity in BRCA1 mutant ovarian tumors. The combination of rucaparib and either an anti-PD-L1 or anti-CTLA-4 antibody were equally compelling in preclinical studies.
ATHENA is a four-arm first-line maintenance treatment study to evaluate Rubraca and Opdivo, Rubraca, Opdivo and placebo in an estimated 1,000 newly diagnosed patients with stage III/IV high-grade ovarian, fallopian tube, or primary peritoneal cancer who have completed platinum-based chemotherapy. The primary objectives are to determine if the combination of Rubraca and Opdivo meaningfully extends PFS versus Rubraca monotherapy, or versus placebo, and to determine if Rubraca extends PFS versus placebo. The analysis of the study results will evaluate, in a step-down manner, the tBRCA/HRD and intent-to-treat subpopulations. Clovis Oncology is the sponsor, and will also conduct and fund the ATHENA study, which is expected to initiate in the first half of 2018.
The combination study of Rubraca and Opdivo in patients with advanced TNBC will be sponsored and conducted by BMS, with study costs to be shared by Clovis and BMS. The study is expected to initiate in the first half of 2018.
The mCRPC study is a three-arm study evaluating Opdivo + Rubraca, Opdivo + docetaxel + prednisone, and Opdivo + enzalutamide, with the objective of determining how the combination affects objective response rate and PSA response. The study will enroll patients with biomarker negative or positive disease, and tumor tissue samples will be used to determine biomarker status. BMS is sponsoring, conducting and funding the 300 patient study in mCRPC, which initiated in the fourth quarter of 2017.
In addition, a Phase 1b study (WO39409; NCT NCT03101280) sponsored by Hoffman-La Roche (Genentech) is underway evaluating the combination of cancer immunotherapy Tecentriq® (atezolizumab; anti-PDL1) and Rubraca for the treatment of advanced gynecological cancers and TNBC in patients with a tumor BRCA or HRD mutation. This study began enrolling patients in the first half of 2017.
Companion Diagnostics
The company partnered with Foundation Medicine, Inc. to co-develop a companion diagnostic test, the FDA approved FoundationFocus™ CDx BRCA, to select patients for treatment with Rubraca in the ovarian cancer treatment indication. FoundationFocus CDx BRCA is a next-generation sequencing (“NGS”) assay that assesses tumor BRCA mutations from tumor tissue samples from patients with ovarian cancer.
On November 30, 2017, Foundation announced FDA approval of its comprehensive companion diagnostic test for solid tumors, FoundationOne™ CDx, a NGS based in vitro diagnostic device for detection of substitutions, insertion and deletion alterations, and copy number alterations in 324 genes and select gene rearrangements, as well as genomic signatures including microsatellite instability and tumor mutational burden. Among the genes assessed by the test are BRCA1/2, and FoundationOne™ CDx is approved as a companion diagnostic to select patients for treatment with Rubraca in the advanced ovarian cancer treatment indication.
As part of its collaboration with Foundation, the company also developed an NGS test to use as a diagnostic to assess genomic LOH in tumor samples, and this biomarker has the potential to expand the clinical utility of Rubraca in ovarian cancer and other indications. This test, FoundationFocus™ CDx BRCA HRD test, was used to assess HRD status in patient tumor samples in the ARIEL3 study.
The company also have a companion diagnostic collaboration with Myriad Genetics, Inc. (“Myriad”) to support a post-marketing regulatory commitment related to Rubraca’s initial approval in the ovarian cancer treatment setting. Myriad will submit a supplementary premarket approval (“sPMA”) application under its existing PMA for BRACAnalysis CDx to include Rubraca. BRACAnalysis CDx is Myriad’s blood-based assay for the qualitative detection and classification of germline mutations in BRCA1/2 genes.
Lucitanib – a VEGFR, PDGFR and FGFR Inhibitor
Lucitanib is an oral inhibitor of the tyrosine kinase activity of vascular endothelial growth factor receptors (VEGFR) 1-3, platelet-derived growth factor receptors (PDGFR) alpha and beta and fibroblast growth factor receptors (FGFR) 1-3. Lucitanib was previously evaluated in breast and lung cancers. Development in those indications has ceased and the company continue to provide drug to patients whose clinicians recommend continuing lucitanib therapy. Clovis Oncology is continuing to evaluate what, if any, further development of lucitanib will be pursued. The company hold development and commercialization rights in the U.S. and Japan and have sublicensed rights to Europe and rest of world markets, excluding China, to Servier.
Rociletinib - an Oral EGFR Mutant-Selective Inhibitor
Rociletinib is an oral mutant-selective inhibitor of epidermal growth factor receptor (“EGFR”). The company terminated enrollment in all sponsored clinical studies, although the company continue to provide drug to patients whose clinicians recommend continuing rociletinib therapy. Clovis Oncology is continuing analyses of rociletinib data to determine whether certain populations of patients may represent an opportunity for a partner committed to investing in further clinical development. The company hold global development and commercialization rights for rociletinib.
Competition
The development and commercialization of new drugs is competitive, and the company face competition from major pharmaceutical and biotechnology companies worldwide. The company's competitors may develop or market products or other novel technologies that are more effective, safer or less costly than any that have been or will be commercialized by it, or may obtain regulatory approval for their products more rapidly than the company may obtain approval for it.
The acquisition or licensing of pharmaceutical products is also very competitive, and a number of more established companies, which have acknowledged strategies to license or acquire products, may have competitive advantages over it, as may other emerging companies taking similar or different approaches to product acquisitions. Many of its competitors have substantially greater financial, technical and human resources than Clovis Oncology has. Additional mergers and acquisitions in the pharmaceutical industry may result in even more resources being concentrated in its competitors. Competition may increase further as a result of advances made in the commercial applicability of technologies and greater availability of capital for investment in these fields. The company's success will be based in part on its ability to build and actively manage a portfolio of drugs that addresses unmet medical needs and creates value in patient therapy.
Rubraca Competition
Lynparza®/olaparib (AstraZeneca) was the first PARP inhibitor to market and has been approved in the US in the following indications:
as monotherapy for the treatment of adult patients who have deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy;as monotherapy for the maintenance treatment of adult patients with recurrent epithelial, fallopian tube or primary peritoneal cancer, who are in a complete or partial response to platinum-based chemotherapy; andas monotherapy in patients with deleterious or suspected deleterious gBRCAm, human epidermal growth factor 2 (HER2)-negative metastatic breast cancer who have been treated with chemotherapy in the neoadjuvant, adjuvant, or metastatic setting.
Lynparza is approved in the EU for use as monotherapy for the maintenance treatment of adult patients with platinum-sensitive relapsed BRCA-mutated (germline and/or somatic) high grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in response (complete response or partial response) to platinum-based chemotherapy. In February 2018, the CHMP adopted a positive opinion recommending this label be changed and expanded to remove the limitation of use in BRCA-mutated patients. Lynparza has indications for ovarian cancer across 57 countries (as of year-end 2017).
In July 2017, AstraZeneca and Merck & Co., Inc. announced a global strategic oncology collaboration to co-develop and co-commercialise Lynparza for multiple cancer types. Lynparza is being investigated, alone and in combination with other agents, in multiple indications across several tumor types, including breast, prostate, and pancreatic cancers.
Zejula®/niraparib (Tesaro) was approved in March 2017 in the United States as monotherapy for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy. Zejula was approved in November 2017 in the EU for the same indication. Additional clinical investigations of Zejula in ovarian, breast and lung cancers are ongoing or planned. Janssen Biotech has licensed rights to develop and commercialize niraparib specifically for patients with prostate cancer worldwide, except in Japan.
There are a number of PARP inhibitors in clinical development including AbbVie’s veliparib and ABT-767, Pfizer’s talazoparib, BeiGene’s BGB-290, and Checkpoint Therapeutics’ CK-102. While most PARP inhibitor development focuses on ovarian, breast and prostate cancers, additional efforts are aimed toward bladder, lung, and pancreatic cancers as well.
In addition, combination approaches that include PARP inhibitors, including Lynparza or Zejula, with other anticancer agents are in various phases of clinical development across a variety of oncology indications. These combination therapies may result in future competitive pressure on Rubraca, and multiple data readouts for such studies are anticipated throughout 2018 and beyond.
Outside of the PARP class, Avastin®/bevacizumab is approved in the US for recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer that is platinum-resistant in combination with paclitaxel, pegylated liposomal doxorubicin, or topotecan, and was approved in December 2016 in the US for recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer that is platinum-sensitive in combination with carboplatin and paclitaxel or in combination with carboplatin and gemcitabine, followed by Avastin as a single agent. Other out of class agents approved for use in advanced ovarian cancer include chemotherapeutic agents (e.g. platinum-based doublets, platinum monotherapy, non-platinum chemotherapy, etc.), Doxil® (Janssen), and Hycamtin® (Novartis). There are additional out-of-class agents in clinical development that may pose a future competitive threat to Rubraca.
License Agreements
Pfizer Inc.
In June 2011, the company entered into a license agreement with Pfizer Inc. (“Pfizer”) to obtain the exclusive global rights to develop and commercialize Rubraca. The exclusive rights are exclusive even as to Pfizer and include the right to grant sublicenses. Pursuant to the terms of the license agreement, the company made a $7.0 million upfront payment to Pfizer and are required to make additional payments to Pfizer for the achievement of certain development and regulatory and sales milestones and royalties on sales as required by the license agreement. Prior to the FDA approval of Rubraca, the company made milestones payments of $1.4 million, which were recognized as acquired in-process research and development expense.
On August 30, 2016, the company entered into a first amendment to the worldwide license agreement with Pfizer, which amends the June 2011 existing worldwide license agreement to permit it to defer payment of the milestone payments payable upon ![]() FDA approval of an NDA for 1st Indication in US and (ii) European Commission approval of an MAA for 1st Indication in EU, to a date that is 18 months after the date of achievement of such milestones. In the event that the company defer such milestone payments, Clovis Oncology has agreed to pay an additional $3.0 million related to the achievement of each such milestone.
FDA approval of an NDA for 1st Indication in US and (ii) European Commission approval of an MAA for 1st Indication in EU, to a date that is 18 months after the date of achievement of such milestones. In the event that the company defer such milestone payments, Clovis Oncology has agreed to pay an additional $3.0 million related to the achievement of each such milestone.
On December 19, 2016, the FDA approved Rubraca as monotherapy for the treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer, who have been treated with two or more chemotherapies, and selected for therapy based on an FDA-approved companion diagnostic for Rubraca. The FDA approval resulted in a $0.75 million milestone payment to Pfizer as required by the license agreement, which was made in the first quarter of 2017. The FDA approval also resulted in the obligation to pay a $20.0 million milestone payment, for which Clovis Oncology has exercised the option to defer payment by agreeing to pay $23.0 million within 18 months after the date of the FDA approval. These payments were recognized as intangible assets and amortized over the estimated remaining useful life of Rubraca.
Clovis Oncology is obligated under the license agreement to use commercially reasonable efforts to develop and commercialize Rubraca and Clovis Oncology is responsible for all ongoing development and commercialization costs for Rubraca. Clovis Oncology is required to make regulatory milestone payments to Pfizer of up to an additional $69.75 million in aggregate if specified clinical study objectives and regulatory filings, acceptances and approvals are achieved. In addition, Clovis Oncology is obligated to make sales milestone payments to Pfizer if specified annual sales targets for Rubraca are met, which relate to annual sales targets of $250.0 million and above, which, in the aggregate, could amount to total milestone payments of $170.0 million, and tiered royalty payments at a mid-teen percentage rate on its net sales, with standard provisions for royalty offsets to the extent the company need to obtain any rights from third parties to commercialize Rubraca.
The license agreement with Pfizer will remain in effect until the expiration of all of its royalty and sublicense revenue obligations to Pfizer, determined on a product-by-product and country-by-country basis, unless the company elect to terminate the license agreement earlier. If the company fail to meet its obligations under the agreement and are unable to cure such failure within specified time periods, Pfizer can terminate the agreement, resulting in a loss of its rights to Rubraca and an obligation to assign or license to Pfizer any intellectual property rights or other rights the company may have in Rubraca, including its regulatory filings, regulatory approvals, patents and trademarks for Rubraca.
AstraZeneca UK Limited
In April 2012, the company entered into a license agreement with AstraZeneca UK Limited (“AstraZeneca”) to acquire exclusive rights associated with Rubraca under a family of patents and patent applications that claim methods of treating patients with PARP inhibitors in certain indications. The license enables the development and commercialization of Rubraca for the uses claimed by these patents. Pursuant to the terms of the license agreement, the company made an upfront payment of $0.25 million upon execution of the agreement. During the second quarter of 2016, the company made a milestone payment of $0.3 million to AstraZeneca upon the NDA submission for Rubraca. These payments were recognized as acquired in-process research and development expense. The FDA approval of Rubraca on December 19, 2016 resulted in a final $0.35 million milestone payment to AstraZeneca as required by the license agreement. This payment was recognized as intangible assets and amortized over the estimated remaining useful life of Rubraca. AstraZeneca also receives royalties on net sales of Rubraca.
Advenchen Laboratories LLC
In October 2008, Ethical Oncology Science, S.p.A. (“EOS”) (now known as Clovis Oncology Italy S.r.l.) entered into an exclusive license agreement with Advenchen Laboratories LLC (“Advenchen”) to develop and commercialize lucitanib on a global basis, excluding China. Clovis Oncology is obligated to pay Advenchen tiered royalties at percentage rates in the mid-single digits on net sales of lucitanib, based on the volume of annual net sales achieved. In addition, after giving effect to the first and second amendments to the license agreement, Clovis Oncology is required to pay to Advenchen 25% of any consideration, excluding royalties, the company receive from sublicensees, in lieu of the milestone obligations set forth in the agreement. Clovis Oncology is obligated under the agreement to use commercially reasonable efforts to develop and commercialize at least one product containing lucitanib, and Clovis Oncology is also responsible for all remaining development and commercialization costs for lucitanib.
The license agreement with Advenchen will remain in effect until the expiration of all of its royalty obligations to Advenchen, determined on a product-by-product and country-by-country basis, unless the company elect to terminate the agreement earlier. If the company fail to meet its obligations under the agreement and are unable to cure such failure within specified time periods, Advenchen can terminate the agreement, resulting in a loss of its rights to lucitanib.
Les Laboratoires Servier
In September 2012, EOS entered into a collaboration and license agreement with Les Laboratoires Servier and Institut de Recherches Internationales Servier (collectively, “Servier”), whereby EOS sublicensed to Servier exclusive rights to develop and commercialize lucitanib in all countries outside of the U.S., Japan and China. In exchange for these rights, EOS received an upfront payment of €45.0 million. Clovis Oncology is entitled to receive additional payments on the achievement of specified development, regulatory and commercial milestones up to €100.0 million in the aggregate, €10.0 million of which was received in the first quarter of 2014. In addition, Clovis Oncology is entitled to receive sales milestone payments if specified annual sales targets for lucitanib are met, each of which relates to annual sales targets of €250.0 million and above, which, in the aggregate, could amount to a total of €250.0 million. Clovis Oncology is also entitled to receive royalties at percentage rates ranging from low-to-mid teens on sales of lucitanib by Servier.
The company and Servier are developing lucitanib pursuant to a development plan agreed to between the parties. Servier is responsible for all of the development costs for lucitanib up to €80.0 million. Cumulative global development costs in excess of €80.0 million, if any, will be shared equally between it and Servier. During the second quarter of 2016, the company and Servier agreed to discontinue the development of lucitanib for breast cancer. During 2017, the company completed the committed on-going development activities and received full reimbursement of its development costs from Servier. Reimbursements are recorded as reduction to research and development expense on the Consolidated Statement of Operations.
The collaboration and license agreement will remain in effect until the expiration of all of Servier’s royalty obligations to it, determined on a product-by-product and country-by-country basis, unless Servier elects to terminate the agreement earlier. If the company fail to meet its obligations under the agreement and are unable to cure such failure within specified time periods, Servier can terminate the agreement, resulting in the granting of a perpetual license to Servier of rights to lucitanib.
Celgene Corporation
In May 2010, the company entered into an exclusive worldwide license agreement with Avila Therapeutics, Inc. (now Celgene Avilomics Research Inc., part of Celgene Corporation (“Celgene”)) to discover, develop and commercialize a covalent inhibitor of mutant forms of the EGFR gene product. Rociletinib was identified as the lead inhibitor candidate under the license agreement. Clovis Oncology is responsible for all non-clinical, clinical, regulatory and other activities necessary to develop and commercialize rociletinib.
The company made an upfront payment of $2.0 million upon execution of the license agreement, a $4.0 million milestone payment in the first quarter of 2012 upon the acceptance by the FDA of its Investigational New Drug (“IND”) application for rociletinib and a $5.0 million milestone payment in the first quarter of 2014 upon the initiation of the Phase II study for rociletinib. In the third quarter of 2015, the company made milestone payments totaling $12.0 million upon acceptance of the NDA and MAA for rociletinib by the FDA and EMA, respectively. The company recognized all payments prior to commercial approval as acquired in-process research and development expense.
Clovis Oncology is obligated to pay royalties at percentage rates ranging from mid-single digits to low teens on the volume of annual net sales achieved. Clovis Oncology is required to pay up to an additional aggregate of $98.0 million in development and regulatory milestone payments if certain clinical study objectives and regulatory filings, acceptances and approvals are achieved. In addition, Clovis Oncology is required to pay up to an aggregate of $120.0 million in sales milestone payments if certain annual sales targets are achieved.
Clovis Oncology has full sublicensing rights under the license agreement, subject to its sharing equally with Celgene any upfront payments from any sub-licensing arrangements relating to Japan, or Japan and any one or more of China, South Korea and Taiwan, which the company refer to herein as an Asian Partnership, and subject to its paying royalties on sales in Asia equal to the greater of the royalty rates contained in its license agreement or 50% of the royalties the company receive from its Asian Partnership.
The license agreement will remain in effect until the expiration of all of its royalty and sublicense revenue obligations to Celgene, determined on a product-by-product and country-by-country basis, unless the company elect to terminate the license agreement earlier. If the company fail to meet its obligations under the agreement and are unable to cure such failure within specified time periods, Celgene can terminate the agreement, resulting in a loss of its rights to rociletinib and an obligation to assign or license to Celgene any intellectual property rights or other rights the company may have in rociletinib, including its regulatory filings, regulatory approvals, patents and trademarks for rociletinib.




