Global Blood Therapeutics
Overview
Global Blood Therapeutics (GBT) is a clinical-stage biopharmaceutical company determined to discover, develop and deliver innovative treatments that provide hope to underserved patient communities. The company's lead product candidate is voxelotor (previously known as GBT440), an oral, once-daily therapy that modulates hemoglobin’s affinity for oxygen, which the company believe inhibits hemoglobin polymerization in sickle cell disease, or SCD.1
Global Blood Therapeutics is currently evaluating voxelotor in SCD in a Phase 3 clinical trial in adult and adolescent patients with SCD. In addition, Global Blood Therapeutics is evaluating the safety and pharmacokinetics of single and multiple doses of voxelotor in a Phase 2a clinical trial of adolescent and pediatric patients with SCD, and in July 2017 the company announced that Global Blood Therapeutics has expanded this open-label trial to include a new single-dose cohort in children aged 6-11. In December 2015, the Food and Drug Administration, or FDA, granted Fast Track Designation and Orphan Drug Designation for voxelotor for the treatment of SCD and in November 2016 voxelotor was granted Orphan Drug Designation in Europe for the treatment of SCD. In June 2017, the European Medicines Agency, or EMA, granted PRIME designation for voxelotor for the treatment of SCD. The PRIME program is a new regulatory mechanism that provides for early and proactive EMA support to medicine developers to help patients benefit as early as possible from innovative new products that have demonstrated the potential to significantly address an unmet medical need. In September 2017, the FDA granted Rare Pediatric Disease designation to voxelotor for the treatment of SCD. In January 2018, the FDA granted Breakthrough Therapy Designation to voxelotor for the treatment of SCD.
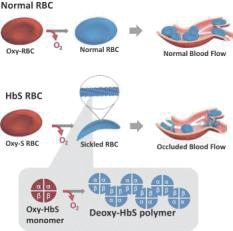
SCD is marked by red blood cell, or RBC, destruction and occluded blood flow and hypoxia, leading to anemia, stroke, multi-organ failure, severe pain crises, and shortened patient life span. Voxelotor inhibits abnormal hemoglobin polymerization, the underlying mechanism that causes sickling of RBCs. In its clinical trials to date of voxelotor in SCD patients, the company observed reduced markers of red blood cell destruction, improvements in anemia, improvements in markers of tissue oxygenation, and reduced numbers of sickled RBCs.
SCD is a genetic blood disorder caused by a single point mutation in the beta-chain of hemoglobin, which results in the formation of abnormal hemoglobin known as sickle hemoglobin, or HbS. Normally, oxygenated RBCs travel from the lung through blood vessels. Hemoglobin is the protein inside RBCs that carries oxygen and releases oxygen at the tissues. In SCD, when oxygen is released, HbS becomes sticky and aggregates into polymers, or long, rigid rods within RBC, much like a “sword within a balloon.” The RBCs assume a sickled shape and becomes inflexible, which can destroy RBCs, cause blockage in small blood vessels, block blood flow, and decrease oxygen delivery to tissues. As a result, beginning in early childhood, SCD patients suffer many clinical consequences, including unpredictable and recurrent episodes, or crises, of severe chronic and acute pain, anemia, stroke, spleen failure, pulmonary hypertension, acute chest syndrome, liver disease, kidney failure, other morbidities, and premature death. These consequences are directly related to reduced blood flow and insufficient oxygen delivery. According to a 2014 publication, in the United States, SCD shortens patient life expectancy by approximately 25 to 30 years even with available medical care.
Current treatment options for SCD are limited to one approved therapy for adults known as hydroxyurea, one recently approved treatment for patients age five years and older called L-glutamine (marketed as EndariTM), blood transfusions and bone marrow transplantation. The utilization of these treatments is significantly limited, for hydroxyurea due to suboptimal efficacy and significant toxicity. As a result, patients with SCD continue to suffer serious morbidity and premature mortality.
The company believe there is a significant unmet medical need for a novel SCD therapy that:
- inhibits abnormal hemoglobin polymer formation, the underlying mechanism of RBC sickling;
- stops inappropriate RBC destruction and improves blood flow and oxygen delivery to tissues;
- prevents or reduces the episodes or crises of severe pain associated with SCD;
- modifies the long-term course of the disease;
- is effective in all SCD genotypes, and in both children and adults;
- has a more favorable side effect profile than currently available therapies; and
- is available as a convenient, oral therapy.
Voxelotor’s therapeutic approach was inspired by the natural activity of fetal hemoglobin, or HbF. HbF is present during fetal development and in early infancy until it is replaced with adult hemoglobin, and has an inherently increased oxygen affinity that allows a fetus to extract oxygen from the mother’s blood. Typically, newborns with SCD do not experience RBC sickling until approximately six to nine months of age, after which HbF is usually no longer expressed. Additionally, it has been observed that rare individuals who have inherited both the HbS mutation as well as a gene deletion that allows them to continue to express 10% to 30% HbF in their RBCs into adulthood do not exhibit the clinical manifestations of SCD, despite expressing up to 90% sickled hemoglobin, or HbS in their blood. HbF dilutes the concentration of deoxygenated HbS that can participate in hemoglobin polymerization, and thereby significantly reduces the formation of hemoglobin polymers.
Voxelotor is a novel, proprietary investigational drug that increases hemoglobin’s affinity for oxygen by binding to the alpha-chain of hemoglobin. Voxelotor has been observed to keep a proportion of HbS in its oxygenated state so it cannot participate in polymerization. Similar to HbF, by diluting total HbS with a proportion of voxelotor-bound hemoglobin, voxelotor prevents hemoglobin polymer formation. Based on its mechanism of action, the company believe that voxelotor can reduce the physical and clinical manifestations of SCD.
In December 2014, the company initiated its randomized, placebo-controlled, double-blind, single and multiple ascending dose Phase 1/2 clinical study of voxelotor in healthy subjects and patients with SCD. The study was conducted in three parts: single dose administration, followed by multiple dose administration, daily for 15 days in healthy subjects and 28 days in SCD subjects, and then followed by multiple dose administration, daily for up to six months in SCD subjects. In this clinical trial, the company evaluated the safety, tolerability, pharmacokinetics, and pharmacodynamics of voxelotor, as well as exploratory markers of SCD activity, including anti-hemolytic effects and SCD-related clinical effects. Among the 41 SCD subjects who received multiple doses of voxelotor (at doses of either 1000mg, 900mg, 700mg or 500mg per day) for 28 days up to six months, 100% of study subjects demonstrated a hematologic response to voxelotor therapy, as evidenced by improvements in one or more markers of hemolysis and anemia (hemoglobin, unconjugated bilirubin and/or percentage reticulocyte counts). In addition, all study cohorts showed marked reductions in irreversibly sickled cells in the peripheral blood from baseline, and, all study subjects treated for 90 days to up to six months showed sustained improvement in bilirubin and/or percentage reticulocyte counts, continued reductions in sickled RBCs and a median 1.0 g/dL increase in hemoglobin. In this clinical study, voxelotor was well tolerated through six months of dosing; the most common treatment-related adverse events were mild to moderate headaches and gastrointestinal disorders, which occurred in similar rates in the placebo arms.
In August 2016, the company initiated its open-label, single- and multiple-dose Phase 2a clinical study of voxelotor in adolescent and pediatric patients with SCD that the company refer to as the HOPE-KIDS 1 Study. In July 2017, the company expanded this open-label trial to include a single-dose cohort in children aged 6-11. The HOPE-KIDS 1 Study is designed to evaluate the safety, tolerability, pharmacokinetics and exploratory treatment effect of voxelotor in a pediatric population (age 6 to 17) with SCD. The study is being conducted in two parts: the single-dose Part A portion includes two cohorts of patients who will receive a single oral dose of 600 mg of voxelotor. Cohort 1 is complete, with seven patients age 12 to 17 enrolled. Cohort 2 is complete with six patients age 6 to 11 enrolled. Part B will explore the safety of multiple doses of voxelotor administered to patients age 12 to 17 for 24 weeks. The doses being evaluated, 900 mg per day and 1500 mg per day, are consistent with those currently being administered in its ongoing Phase 3 clinical trial of voxelotor in adult and adolescent patients with SCD. Among the 13 SCD patients who received a single oral dose of 600 mg of voxelotor, 100% of the study patients demonstrated that voxelotor was well tolerated, with no serious or severe adverse events related to study drug observed. In addition, the pharmacokinetics and half-life of voxelotor were similar in adolescents (age 12 to 17) and adults, with results supporting once-daily dosing and a high specificity for hemoglobin. Among the 11 SCD adolescent patients who received multiple doses of voxelotor at 900 mg per day for whom data were available at 16 weeks, 55 percent of patients demonstrated a hematologic response to voxelotor therapy, as evidenced by improvements in one or more markers of hemolysis and anemia (hemoglobin, unconjugated bilirubin and percentage reticulocyte counts). In this clinical study, voxelotor was well tolerated; the most common treatment-related adverse events were mild to moderate headaches, nausea and rash.
Following discussions with the FDA that concluded successfully in October 2016, Global Blood Therapeutics has initiated a randomized, double-blind, placebo-controlled, multi-national Phase 3 clinical trial of voxelotor in SCD that the company refer to as the HOPE Study. The HOPE Study is designed to enroll up to approximately 400 adult and adolescent SCD patients, age 12 years and older, who have had at least one episode of vaso-occlusive crisis, or VOCs, in the previous year. The HOPE Study is being conducted in two parts: the initial Part A, which compares two dose levels of voxelotor (900 mg and 1500 mg) versus placebo, and will include up to 150 patients; followed by Part B, which will include 250 patients randomized to placebo or a dose of voxelotor selected based on the results of Part A. The main objectives of Part A are to select the optimal dose, define the final secondary endpoints for Part B and qualify the Patient Reported Outcome, or PRO, instrument. The primary efficacy endpoint of the HOPE Study is the proportion of patients who achieve a >1 g/dL increase in hemoglobin at 24 weeks of treatment compared to baseline. Key secondary efficacy endpoints include the effect of voxelotor on SCD symptom exacerbation, which will be measured by its PRO instrument, in addition to overall SCD symptoms as compared to placebo. The company will also assess traditionally defined VOC as well as hospitalizations and red blood cell transfusions as secondary endpoints.
The company believe there is a significant market opportunity in SCD. The U.S. Centers for Disease Control, or CDC, estimates the prevalence of SCD at approximately 100,000 individuals in the United States, where newborn screening is mandatory. It is estimated that the prevalence of SCD in Europe is approximately 60,000 individuals. The global incidence of SCD is estimated to be 250,000 to 300,000 births annually. One study estimates that in the United States, the average annual cost for the care of an adult patient with the most common genotype of SCD exceeds $200,000, and the cumulative lifetime costs exceed $8.0 million over an assumed 50-year lifespan, driven primarily by hospital admissions, physician fees, clinic and emergency department visits, and the costs of diagnostic procedures and outpatient consultations.
The company own or jointly own and have exclusively licensed rights to its product candidates in the United States, Europe and other major markets. Global Blood Therapeutics is the sole owner of issued U.S. patents covering voxelotor, including its composition of matter, methods of use, and a polymorph of voxelotor. These issued patents covering voxelotor will expire between 2032 and 2035, absent any applicable patent term extensions. The company own or co-own additional pending patent applications in the United States and multiple foreign countries relating to its lead product candidate voxelotor.
To execute on the opportunities presented by its research and development portfolio, Global Blood Therapeutics has assembled a team of employees, management and directors rich in scientific experience and capabilities in drug discovery, development and commercialization. The company's management has a successful track record in developing and commercializing drug candidates. In aggregate, its management team has contributed to several drug approvals, including Avastin®, Herceptin®, Kaletra®, Kyprolis®, Ravicti® and Rituxan®. The company intend to leverage this expertise and experience to rapidly advance the development of voxelotor for SCD and advance other product candidates. Beyond evaluating voxelotor in SCD, Global Blood Therapeutics is also engaged in other research and development activities, all of which are currently in the pre-clinical phase. In addition, the company regularly evaluate opportunities to in-license, acquire or invest in new business, technology or assets or engage in related discussions with other business entities.
Development Pipeline
The following table summarizes its development programs, potential indications, and their current stages of development:

Global Blood Therapeutics was previously conducting two Phase 2a clinical trials of voxelotor for the potential treatment of idiopathic pulmonary fibrosis (IPF), which is a hypoxemic pulmonary disorder. The company also conducted a Phase 1 study called Basecamp in healthy volunteers, which was intended to support its understanding of voxelotor’s effects on hypoxemia and complement its Phase 2a program in IPF. In October 2017, based upon the totality of the data the company obtained from the two Phase 2a clinical studies and Basecamp study the company decided to halt further development of voxelotor for the potential treatment of IPF. While the results re-affirmed its confidence in the mechanism of action of voxelotor in SCD, the data from the IPF proof-of-concept studies did not demonstrate sufficient overall clinical benefit to justify continuing the program in IPF.
Voxelotor for the Treatment of Sickle Cell Disease
Global Blood Therapeutics is developing its lead product candidate voxelotor as a once-daily, oral therapy for patients with SCD. Global Blood Therapeutics is investigating voxelotor’s potential to inhibit the abnormal polymerization of hemoglobin, which is the underlying mechanism of RBC, sickling and leads to the associated complications that characterize SCD. Global Blood Therapeutics has designed a clinical program for voxelotor targeted at the treatment of adults, adolescents, children and infants across all SCD genotypes. In December 2014, the company initiated its first clinical trial of voxelotor, a Phase 1/2 study in which the company evaluated voxelotor in both healthy subjects and SCD subjects. In November 2016, the company initiated its on-going multi-national Phase 3 clinical trial of voxelotor in adult and adolescent subjects with SCD, which Global Blood Therapeutics has named the HOPE Study. In addition, Global Blood Therapeutics is evaluating the safety and pharmacokinetics of single and multiple doses of voxelotor in a Phase 2a clinical trial of adolescent and pediatric patients with SCD, and in July 2017 the company announced that Global Blood Therapeutics has expanded this open-label trial to include a new single-dose cohort in children aged 6-11.
Sickle Cell Disease Overview
SCD is a grievous disease that can lead to hemolytic anemia, meaning the destruction of RBCs within blood vessels, and vaso-occlusion, which means blocked blood flow to tissues, as well as progressive multi-organ damage and early death. Beginning in childhood, patients suffer unpredictable and recurrent episodes or crises of severe pain due to blocked blood flow to organs, which often lead to physical and psychosocial disability. In addition, the constant destruction of RBCs with the release of their contents into the blood often leads to damaged or diseased blood vessels, which further exacerbate blood flow obstruction and multi-organ damage. Consequences of SCD can manifest in early childhood and may include stroke, spleen failure, pulmonary hypertension, acute chest syndrome, liver disease, kidney failure, leg ulcers, priapism, which is a medical emergency due to refractory penile erection, and premature death. According to a 2014 publication, in the United States, SCD shortens patient life expectancy by approximately 25 to 30 years even with available medical care.
SCD is a genetic blood disorder caused by a single gene mutation in the beta-chain of hemoglobin, which results in mutant hemoglobin known as HbS. Hemoglobin is the protein in RBCs that carries oxygen from the lungs to the body’s tissues, releases oxygen at the tissues, and returns carbon dioxide from the tissues back to the lungs. Hemoglobin accomplishes this by binding and then releasing oxygen through allosterism, which means the hemoglobin molecule changes its shape to have a high affinity for oxygen in the lungs, where oxygen is abundant, and to have a low affinity for oxygen in the tissues, where oxygen must be released. Oxyhemoglobin, the high oxygen affinity form of hemoglobin, is formed in the lungs during respiration, when oxygen binds to the hemoglobin molecule. Deoxyhemoglobin, the low oxygen affinity form of hemoglobin, is formed when oxygen molecules are removed from the binding site as blood flows from the lungs to the tissues in the body. In patients with SCD, deoxygenated HbS, molecules polymerize to form long, rigid rods within an RBC, much like a “sword within a balloon.” As a consequence, the normally round and flexible RBC becomes rigid and elongate into a “sickled” shape. Sickled RBCs do not flow properly in the bloodstream; they clog small blood vessels and reduce blood flow to the organs. This results in inadequate oxygen delivery, or hypoxia, to all body tissues, which can lead to multi-organ failure and premature death.
The following graphic illustrates the process by which sickling occurs in SCD patients as a result of the polymerization of deoxygenated HbS in an RBC, leading to occluded blood flow, in contrast to a normal RBC:
SCD manifests in individuals who inherit at least one HbS gene from a parent and an additional mutation on the second beta globin gene from the other parent. There are several different genotypes of SCD, including the following major genotypes:
- HbSS, or sickle cell anemia, where both genes are HbS;
- HbSC, where one gene is HbS, and the other is HbC (inherited by a non-SCD impacted parent); and
- HbS/ßthal, where one gene is HbS, and the other is Beta thalassemia.
Market Opportunity in SCD
The CDC estimates the prevalence of SCD at approximately 100,000 individuals in the United States, where newborn screening is mandatory. The incidence of SCD is estimated at approximately 1 in 2,000 to 2,500 newborns in the United States. It is estimated that the prevalence of SCD in Europe is approximately 60,000 individuals. The global incidence of SCD is estimated to be 250,000 to 300,000 births annually. SCD is concentrated in populations of African, Middle Eastern and South Asian descent.
Of SCD patients in the United States, approximately 45% are under the age of 18, and approximately 60% to 65% have the HbSS genotype, which is often referred to as sickle cell anemia, with the remaining 35% to 40% having other genotypes. In all genotypes of SCD, the mechanism that leads to the consequences of the disease involves the polymerization of HbS in its deoxygenated state, which results in RBC sickling. The company believe that because of this common underlying mechanism, voxelotor may show activity across all SCD genotypes. The company's Phase 3 HOPE Study is enrolling SCD patients with all genotypes of SCD.
SCD is associated with high treatment costs. One study estimates that in the United States, the average annual cost for the care of an adult patient with the most common genotype of SCD exceeds $200,000, and the cumulative lifetime costs exceed $8.0 million over an assumed 50-year lifespan, driven primarily by hospital admissions, physician fees, clinic and emergency department visits and the costs of diagnostic procedures and outpatient consultations. As a result, the company believe that a safe, effective and convenient oral treatment for SCD would be well received by patients, physicians and payors.
Current Treatment Options and Their Limitations
SCD remains a significant unmet medical need. The first drug approved to treat SCD, known as hydroxyurea, which was initially approved as a chemotherapy drug, was approved by the FDA in 1998 for the treatment of sickle cell anemia in adults with 3 or more painful crises per year. Hydroxyurea is currently the only drug approved for SCD, and it is not approved for pediatric SCD patients in the United States. The use of hydroxyurea is significantly limited by its side effect profile, variable patient responses and concerns regarding long-term toxicity. Hydroxyurea’s side effects include impairment of fertility, suppression of white blood cells, or neutropenia, and suppression of platelets, or thrombocytopenia, which place patients at risk for infection and bleeding. In July 2017, the FDA approved L-glutamine oral powder for patients age five and older with SCD to reduce severe complications associated with the disorder. In January 2018, Emmaus Life Sciences, Inc., the marketer for EndariTM (L-glutamine oral powder) announced that the product is now available to patients.
In addition to hydroxyurea treatment and L-glutamine, transfusions with normal blood are an option to help alleviate anemia, which is a common symptom of SCD, and reduce sickling of RBCs. Blood transfusions have a number of limitations, including the expense of treatment, lack of uniform accessibility and risks ranging from allergic reactions to serious complications such as blood-borne infection and iron overload, which can cause organ damage. The only potentially curative treatment currently available for SCD patients is bone marrow transplantation, which requires a suitable matching donor and carries significant risks, including an approximately 5% mortality rate. Despite the current standard of care, including hydroxyurea, blood transfusion and palliative therapy for acute pain attacks, patients with SCD continue to suffer serious morbidity and premature mortality.
In light of the devastating effects of SCD on patients and the high costs of care for these patients, there is a significant unmet need for a treatment that:
- inhibits abnormal hemoglobin polymer formation, the underlying mechanism of RBC sickling;
- stops inappropriate RBC destruction and improves blood flow and oxygen delivery to tissues;
- prevents or reduces the episodes or crises of severe pain associated with SCD;
- modifies the long-term course of the disease;
- is effective in all SCD genotypes, and in both children and adults;
- has a more favorable side effect profile than currently available therapies; and
- is available as a convenient, oral therapy.
Overview of Hemoglobin Biology and Voxelotor’s Mechanism of Action
As described above, hemoglobin accomplishes transports oxygen from the lungs to the body’s tissues, releases oxygen into the tissues, and returns carbon dioxide from the tissues back to the lungs by changing its shape to be high affinity for oxygen in the lungs, where oxygen is abundant, and low affinity for oxygen in the tissues, where oxygen must be released. An important tool for assessing how readily hemoglobin acquires and binds oxygen in the lungs and releases oxygen into the tissues is the oxygen equilibrium curve, or OEC. The OEC represents the proportion of oxyhemoglobin, measured as the percentage of oxygen saturation (O2 % saturation) on the vertical axis relative to the amount of oxygen dissolved in blood, indicated as the oxygen tension, or partial pressure of oxygen (pO2) measured in millimeters of mercury (mmHg), on the horizontal axis.
Global Blood Therapeutics has demonstrated in preclinical models that its novel hemoglobin modifiers, including voxelotor, bind to hemoglobin, resulting in increased oxygen affinity. The effect of voxelotor on the measured OEC (Oxygen Equilibrium Curve) is a shift of the curve to the left. In other words, at a given prevailing oxygen tension in the blood, Global Blood Therapeutics has observed a higher percentage of oxygen saturation, or a higher proportion of oxyhemoglobin in the blood, following the administration of voxelotor.
In various studies of SCD, scientists have demonstrated that hemoglobin in the oxygenated state is a potent inhibitor of HbS polymerization. Since HbS polymerization occurs in the deoxygenated state, the company believe that increasing the proportion of oxyhemoglobin, or “left-shifting” the OEC, should delay the polymerization of HbS and prevent the sickling of RBCs, which may ameliorate many, of the clinical manifestations of SCD. Importantly, Global Blood Therapeutics is able to measure the proportion of hemoglobin modification (%HbMOD), which is expressed as the percentage of hemoglobin molecules occupied or bound by voxelotor.
HbF, which is present during fetal development and persists for up to six to nine months in infants until it is replaced by adult hemoglobin, has an inherent high affinity for oxygen, which is critical for a developing fetus to capture oxygen from the mother’s blood. Newborns with SCD do not experience RBC sickling until approximately six to nine months of age, after which HbF is no longer expressed. Additionally, it has been observed that rare individuals who have inherited the HbS mutation and a gene deletion that allows them to continue to express 10% to 30% HbF in their RBCs into adulthood do not exhibit the clinical manifestations of SCD, despite expressing up to 90% sickled hemoglobin, or HbS, in their blood. HbF dilutes the concentration of deoxygenated HbS that can participate in polymerization, and thereby prevents hemoglobin polymer from forming.
Based on these observations, the company believe that to delay polymerization of HbS, voxelotor would need to bind to only approximately 10% to 30% of the total hemoglobin in a patient’s blood. One theoretical concern regarding increasing the affinity of hemoglobin for oxygen is that excessive oxygen affinity could prevent hemoglobin from releasing oxygen into the tissues, thus causing hypoxia. However, based on HbF data, its animal toxicology studies, and its clinical studies, the company believe its target modification of the total hemoglobin in a patient’s blood would not adversely compromise oxygen delivery to the tissues.
Phase 1/2 Clinical Trial of Voxelotor
In December 2014, the company initiated its first clinical trial of voxelotor, a randomized, placebo-controlled, double-blind, single and multiple ascending dose study in which the company evaluated the safety, tolerability, pharmacokinetics and pharmacodynamics of voxelotor in both healthy subjects and patients with SCD. The company refer to this Phase 1/2 clinical study as the GBT440-001 study. This clinical study was conducted at Quintiles Drug Research Unit at Guy’s Hospital in London, United Kingdom, and was conducted in three parts, as shown in Figure 1 below: Part A (single dose administration), Part B (multiple dose administration, daily for 15 days in healthy subjects and 28 days in SCD subjects), and Part C (multiple dose administration, daily for 90 days in SCD subjects). An extension study for patients in Part C to dose some patients for up to 6 months is referred to as GBT440-024. The company evaluated voxelotor’s ability to prevent the hemolysis or destruction of RBCs in SCD subjects primarily by measuring the blood levels of hemoglobin, bilirubin and reticulocyte counts. The company also measured LDH as an additional marker of hemolysis; however it is generally more variable and nonspecific because it is released from tissues other than RBCs and does not measure extravascular hemolysis which is the primary hemolytic mechanism in SCD. In this clinical trial, the company also evaluated the effect of voxelotor on morphologic sickling of RBCs. The company believe that findings of decreased hemolysis and anti-sickling activity provide evidence of inhibition of sickle hemoglobin polymerization, which may translate into improved symptoms, reduction in clinical events (such as VOC) and reduced organ damage due to RBC sickling.

Overall, a total of 370 subjects, including 283 healthy subjects (including subjects with renal or hepatic impairment) and 53 adult SCD subjects and 28 adolescent subjects with SCD (12 to 17 years of age), and 6 pediatric subjects with SCD (6 to 11 years of age) have been administered single or multiple doses of voxelotor across 15 GBT-sponsored clinical studies. Subjects received single or multiple doses of voxelotor (up to 15 days in healthy subjects, and up to 180 days in subjects with SCD). The studies included 2 Phase 1/2 studies in healthy subjects and subjects with SCD (GBT440-001 and its extension study GBT440-024), 1 Phase 2a study in pediatric participants with SCD (GBT440-007), and 12 clinical pharmacology studies. The most commonly reported adverse events, or AEs, regardless of treatment causality, across SCD subjects included mild or moderate headache, back pain, pain, diarrhea, pain in extremity and rash. All events of diarrhea were mild. Most of these events resolved without treatment and are easily monitored. Of these events, treatment-emergent adverse event were diarrhea, headache and rash. Some subjects with SCD experienced acute painful sickle cell crisis, which was thought to be related to underlying SCD, and not to voxelotor.
In its Phase 1/2 clinical study, the company had no drug-related serious adverse events, or SAEs, reported. A total of 16 SAEs (12 from adult subjects and 4 from adolescent subjects), each of which were assessed to be not related to study drug, were reported in SCD subjects.
Among the 41 SCD patients who received multiple doses of voxelotor (at doses of either 500 mg, 700 mg, 900 mg, or 1000 mg per day) in its Phase 1/2 clinical study for 28 days up to six months, 100% of study subjects demonstrated a hematologic response to voxelotor therapy, as evidenced by significant improvement in one or more clinical markers of hemolysis and anemia (hemoglobin, unconjugated bilirubin and/or percentage reticulocyte counts). In addition, all study cohorts showed marked reductions in irreversibly sickled RBCs in the peripheral blood from baseline, and all study subjects treated for 90 days up to six months showed sustained improvement in bilirubin and/or percentage of reticulocyte counts, continued reductions in sickled RBCs and a median 1.0g/dL increase in hemoglobin. There was no evidence of tissue hypoxia; trends to erythropoietin reductions and the reduction in reticulocyte counts were consistent with an improvement in tissue oxygen delivery; oxygen delivery to tissues at rest and during exercise was normal.
HOPE – KIDS 1 Study
In August 2016, the company initiated its open-label, single and multiple-dose Phase 2a clinical study of voxelotor in adolescent and pediatric patients with SCD which the company refer to as the HOPE-KIDS 1 Study. As shown below in Figure 3, this ongoing study is being conducted in two parts, Part A: single dose of voxelotor at 600 mg in pediatric patients (6 to 11 years) and adolescents (12 to 17 years) and Part B: multiple doses of voxelotor at 2 dose levels, 900 mg/d and 1500 mg/d for 24 weeks in adolescents (approximately 12 patients at each dose). Part A pharmacokinetics, or PK, data in adolescents was previously reported at the 2017 European Hematology Association Congress and demonstrated that the PK and half-life of voxelotor were similar in adolescents and adults with results supporting once-daily dosing. Part A data for pediatric patients (6 to 11 years) was presented at the 2017 American Society of Hematology Annual Meeting, or ASH. Voxelotor PK exposures were higher in children compared with adolescents and adults, which informs dose selection for future pediatric studies in children under 12 years of age. The primary objective of Part B is to assess the effect of voxelotor on anemia. Secondary objectives include effect on clinical measures of hemolysis, PK (PK parameters determined using population PK analysis), daily SCD symptoms as measured by Total Severity Score, or TSS, using a PRO measure and safety.
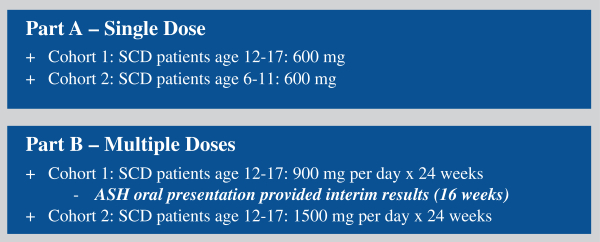
In December 2017 at ASH, the company presented data from the 900 mg Part B portion of the HOPE-KIDS 1 Study for patients that had received 16 weeks of voxelotor treatment. The median age of the patients was 13 years, 92% were on hydroxyurea and 59% had no VOCs in the prior year. Despite the majority of patients being on hydroxyurea, 55% of the patients achieved a hemoglobin response greater than 1 g/dl at week 16. Other clinical measures of hemolysis also improved concordantly; median reduction in reticulocytes and unconjugated bilirubin were 11% and 40% respectively, consistent with the results previously reported in SCD adults treated with voxelotor. Additionally, 10 out of 12 patients showed a reduction in TSS as measured by the PRO from baseline to week 16 of treatment with a 94% median reduction in TSS from baseline. From a safety and tolerability perspective, 900 mg of voxelotor was well tolerated up to 24 weeks of dosing. There were no drug related SAEs and the drug-related AEs were deemed mild to moderate with the exception of one grade 3 rash (which did not recur with continued dosing). There were no study discontinuations due to AEs.
Phase 3 HOPE Study of Voxelotor
The HOPE Study is a randomized, double-blind, placebo-controlled, multi-national, Phase 3 clinical trial enrolling up to approximately 400 adult and adolescent SCD patients age 12 and older who have had at least one episode of VOC in the previous year (see Figure 4). The HOPE Study is being conducted in two parts: the initial Part A, which will compare two dose levels of voxelotor (900 mg and 1500 mg) versus placebo, and will include up to 150 patients; followed by Part B, which will include 250 patients randomized to placebo or a dose of voxelotor selected based on the results of Part A. The main objectives of Part A are to select the optimal dose for Part B, define the final secondary endpoints for Part B and qualify the PRO instrument. The primary efficacy endpoint of the HOPE Study is the proportion of patients who achieve a >1 g/dL increase in hemoglobin at 24 weeks of treatment compared to baseline. The company's discussions with the FDA have focused on a path-way to full approval based on the HOPE Study, by meeting the primary and at least one key secondary endpoint, although achieving this outcome will depend in large part on the data and results obtained from the study itself. Key secondary efficacy endpoints include the effect of voxelotor on SCD symptom exacerbation, which will be measured by its PRO instrument, in addition to overall SCD symptoms as compared to placebo. The PRO is administered on a hand-held electronic device, and is designed to capture the full range of daily SCD symptoms. The HOPE Study will also assess traditionally defined VOC as well as hospitalizations and red blood cell transfusions as secondary endpoints.
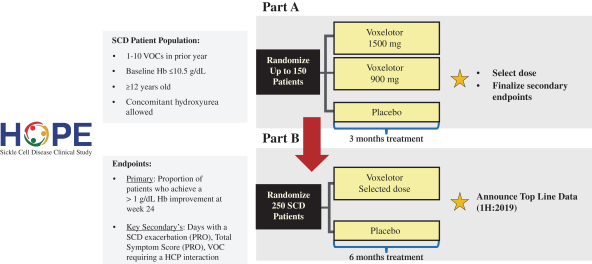
Global Blood Therapeutics is conducting the Phase 3 HOPE Study at leading SCD sites around the globe, enrolling adults and adolescents with SCD who have had at least one episode of VOC in the previous year. The HOPE Study was initiated in December 2016 and the company anticipate announcing top-line Part A results in the first half of 2018 and receipt of top-line data results in the first half of 2019.
European Union Regulatory Path for Voxelotor
Global Blood Therapeutics is engaged in discussions with European regulatory authorities to define the future development plan for voxelotor. The objectives of these regulatory interactions include discussion of study design for additional clinical trials, trial endpoints and the development of voxelotor in other patient populations, including pediatrics.
In November 2016, the European Commission, or EC, granted Orphan Drug Designation status in the European Union, or EU, for voxelotor for the treatment of SCD. In June 2017, the European Medicines Agency, or EMA, granted PRIME designation for voxelotor for the treatment of SCD. The PRIME program is a new regulatory mechanism that provides for early and proactive EMA support to medicine developers to help patients benefit as early as possible from innovative new products that have demonstrated the potential to significantly address an unmet medical need.
Manufacturing
The company do not own or operate, and currently have no plans to establish, any manufacturing facilities. The company currently depend on third-party contract manufacturing organizations, or CMOs, for all of its requirements of raw materials, drug substance and drug product for its nonclinical research and its ongoing clinical trials of its lead product candidate voxelotor. Global Blood Therapeutics has not entered into long-term commercial manufacturing agreements with its current CMOs. The company intend to continue to rely on CMOs for later-stage development and commercialization of voxelotor, as well as the development and commercialization of any other product candidates that the company may identify. Although the company rely on CMOs, Global Blood Therapeutics has personnel and third-party consultants with extensive manufacturing experience to oversee the relationships with its contract manufacturers.
The company believe the synthesis of the drug substance for voxelotor is reliable and reproducible from readily available starting materials, and the synthetic routes are amenable to large-scale production and do not require unusual equipment or handling in the manufacturing process. Global Blood Therapeutics has obtained an adequate supply of the drug substance for voxelotor from its CMOs to satisfy its immediate clinical and nonclinical demands. Global Blood Therapeutics has implemented improvements to its drug substance manufacturing process to further ensure production capacity adequate to meet future development and potential commercial demands.
Global Blood Therapeutics has completed development for a solid oral formulation of a tablet form of voxelotor as well as a pediatric dispersible tablet formulation of voxelotor.
Intellectual Property
The company strive to protect the proprietary technology that the company believe is important to its business, including seeking and maintaining patents and patent applications intended to cover its product candidates and compositions, their methods of use and processes for their manufacture, and any other aspects of inventions that are commercially important to the development of its business. The company also rely on trade secrets to protect aspects of its business that are not amenable to, or that the company do not consider appropriate for, patent protection.
The company plan to continue to expand its intellectual property portfolio. The company endeavor to promptly file domestic and international patent applications for new commercially valuable inventions, including applications directed to compositions and methods of treatment created or identified from its ongoing development of its product candidates. The company's success will depend in part on its ability to obtain and maintain patent and other proprietary rights protecting its commercially important technology, inventions and know-how related to its business, defend and enforce its current and future issued patents, if any, preserve the confidentiality of its trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties. The company also rely on know-how, continuing technological innovation and potential in-licensing opportunities to develop and maintain its intellectual property portfolio.
The patent positions of biopharmaceutical companies like it are generally uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent, if any, is issued, and patent scope can be reinterpreted by the courts after issuance. Moreover, many jurisdictions permit third parties to challenge issued patents in administrative proceedings, which may result in further narrowing or even cancellation of patent claims. The company cannot predict whether the patent applications Global Blood Therapeutics is currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any patents, if issued, will provide sufficient protection from competitors for its business.
Because patent applications in the United States and certain other jurisdictions are maintained in secrecy for 18 months or potentially even longer, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, the company cannot be certain of the priority of inventions covered by pending patent applications. Moreover, the company may have to participate in interference proceedings or derivation proceedings declared by the United States Patent and Trademark Office, or USPTO, to determine the priority of inventions.
Patents
The company's patent portfolio includes multiple issued U.S. patents, as well as multiple U.S. and foreign patent applications in various stages of prosecution or allowance. The company's primary patents and patent applications relate to its general HbS intellectual property portfolio, which includes its lead product candidate voxelotor and its development program.
The company's HbS intellectual property portfolio is comprised of multiple patent families of patents and patent applications relating to voxelotor and/or analogs that inhibit Hb polymerization. These patent families include patents and patent applications specifically related to its lead product candidate voxelotor covering certain compositions of matter, methods of use, method of manufacture, formulations, and polymorphs of voxelotor, as well as certain compositions of matter, methods of use, method of manufacture, formulations, and polymorphs. These patent applications are pending in a variety of jurisdictions, including the United States, jurisdictions under the Patent Cooperation Treaty and other countries.
With regard to voxelotor specifically, Global Blood Therapeutics is the sole owner of issued U.S. patents covering voxelotor, including its composition of matter, methods of use and a polymorph of voxelotor. These issued U.S. patents covering voxelotor will expire between 2032 and 2035, absent any applicable patent term extensions. Any patents that may issue from its pending patent applications relating to voxelotor in the United States or from corresponding foreign patent applications, if issued, are expected to expire between 2032 and 2037, absent any applicable patent term extensions. Some of these pending patent applications are jointly owned by it and Regents of the University of California, or the Regents, as described below.
The company's other patents in its HbS intellectual property portfolio are comprised of additional issued U.S. patents covering voxelotor analogs. These patents, and any patents that may issue from its pending patent applications relating to voxelotor analogs in the United States or from corresponding foreign patent applications, if issued, are currently expected to expire between 2032 and 2034, absent any applicable patent term extensions. Some of these pending patent applications are jointly owned by it and the Regents, as described below.
In addition, Global Blood Therapeutics has exclusively licensed from the Regents worldwide patent rights covering voxelotor and certain voxelotor analogs, some of which patent rights the company jointly own with the Regents. In exchange for its exclusive license, Global Blood Therapeutics has agreed to pay a royalty to the Regents of less than 1% on future net sales and to use commercially reasonable efforts to develop, manufacture, market and sell the products covered by the licensed patents. The risks associated with joint ownership of patent rights are more fully discussed under “Risk Factors-Risks Related to The company's Intellectual Property.”
Beyond its HbS intellectual property portfolio, the company own other issued U.S. patents, seek to obtain additional issued patents, and file patent applications relating to its other research and development programs over time.
Patent term
The base term of a U.S. patent is 20 years from the filing date of the earliest-filed non-provisional patent application from which the patent claims priority, assuming that all maintenance fees are paid. The term of a U.S. patent can be lengthened by patent term adjustment, which compensates the owner of the patent for administrative delays at the USPTO the extent of which is offset by delays by the patent owner before the USPTO in obtaining the patent. In some cases, the term of a U.S. patent is shortened by a terminal disclaimer that reduces its term to that of an earlier-expiring patent. The term of a U.S. patent may be eligible for patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act, to account for at least some of the time the drug is under development and regulatory review after the patent is granted. With regard to a drug for which FDA approval is the first permitted marketing of the active ingredient, the Hatch-Waxman Act allows for extension of the term of one U.S. patent that includes at least one claim covering the composition of matter of an FDA-approved drug, an FDA-approved method of treatment using the drug and/or a method of manufacturing the FDA-approved drug. The extended patent term cannot exceed the shorter of five years beyond the non-extended expiration of the patent or 14 years from the date of the FDA approval of the drug. Some foreign jurisdictions, including Europe and Japan, have analogous patent term extension provisions, which allow for extension of the term of a patent that covers a drug approved by the applicable foreign regulatory agency. In the future, if its lead product candidate voxelotor or any other product candidates receive FDA approval, the company would expect to apply for patent term extension on patents, if issued, covering those products, their methods of use and/or methods of manufacture.




