Halozyme Therapeutics
Halozyme Therapeutics, Inc. (HALO) is a biotechnology company focused on developing and commercializing novel oncology therapies. The company's proprietary enzymes are used to facilitate the delivery of injected drugs and fluids, potentially enhancing the efficacy and the convenience of other drugs or can be used to alter tissue structures for potential clinical benefit. The company exploit its technology and expertise using a two pillar strategy that the company believe enables it to manage risk and cost by: (1) developing its own proprietary products in therapeutic areas with significant unmet medical needs, with a focus on oncology, and (2) licensing its technology to biopharmaceutical companies to collaboratively develop products that combine its technology with the collaborators’ proprietary compounds.1
The majority of its approved product and product candidates are based on rHuPH20, its patented recombinant human hyaluronidase enzyme. The company's proprietary development pipeline consists primarily of pre-clinical and clinical stage product candidates in oncology. The company's lead oncology program is PEGPH20 (PEGylated recombinant human hyaluronidase), a molecular entity Halozyme Therapeutics is developing in combination with currently approved cancer therapies as a candidate for the systemic treatment of tumors that accumulate HA. Halozyme Therapeutics has demonstrated that when HA accumulates in a tumor, it can cause higher pressure in the tumor, reducing blood flow into the tumor and with that, reduced access of cancer therapies to the tumor. Through its efforts and efforts of its partners and collaborators, Halozyme Therapeutics is currently in Phase 3 clinical testing for PEGPH20 with ABRAXANE® (nab-paclitaxel) and gemcitabine in stage IV pancreatic ductal adenocarcinoma (PDA) (Halo 109-301), in Phase 1b clinical testing for PEGPH20 with KEYTRUDA® (pembrolizumab) in non-small cell lung cancer and gastric cancer (Halo 107-101), in Phase 1b/2 clinical testing for PEGPH20 with HALAVEN® (eribulin) in patients treated with up to two lines of prior therapy for HER2-negative metastatic breast cancer, in Phase 1b/2 clinical testing for PEGPH20 with Tecentriq® (atezolizumab) in patients with previously treated metastatic PDA, in Phase 1b/2 clinical testing for PEGPH20 with Tecentriq in patients with gastric cancer and in Phase 1b/2 clinical testing for PEGPH20 with Tecentriq in patients with cholangiocarcinoma and gall bladder cancer (Halo 110-101/MATRIX).
The company refer to the application of rHuPH20 to facilitate the delivery of other drugs or fluids as its ENHANZE™ Technology. The company license the ENHANZE Technology to form collaborations with biopharmaceutical companies that develop or market drugs requiring or benefiting from injection via the subcutaneous route of administration. The company currently have ENHANZE collaborations with F. Hoffmann-La Roche, Ltd. and Hoffmann-La Roche, Inc. (Roche), Baxalta US Inc. and Baxalta GmbH (Baxalta Incorporated was acquired by Shire plc in June 2016) (Baxalta), Pfizer Inc. (Pfizer), Janssen Biotech, Inc. (Janssen), AbbVie, Inc. (AbbVie), Eli Lilly and Company (Lilly) and Bristol-Myers Squibb Company (BMS). The company receive royalties from two of these collaborations, including royalties from sales of one product from the Baxalta collaboration and two products from the Roche collaboration. Future potential revenues from the sales and/or royalties of its approved products, product candidates, and ENHANZE collaborations will depend on the ability of Halozyme and its collaborators to develop, manufacture, secure and maintain regulatory approvals for approved products and product candidates and commercialize product candidates.
third quarter of 2017 and recent highlights include:
- In October 2017, Janssen initiated a Phase 3 study of daratumumab combined with the ENHANZE Technology in amyloidosis patients, with additional Phase 3 studies in multiple myeloma and smoldering myeloma patients planned to initiate in the near term. The company will receive a $15.0 million milestone payment from Janssen upon dosing of the third patient in the recently initiated study.
- In October 2017, Genentech (a member of the Roche Group) initiated a Phase 1b/2 clinical trial evaluating PEGPH20 in combination with Tecentriq in patients with gastric cancer.
- In October 2017, the company initiated the second study in its clinical agreement with Genentech, a Phase 1b/2 open-label randomized study to assess Tecentriq in combination with PEGPH20 and chemotherapy in patients with cholangiocarcinoma and gall bladder cancer.
- In September 2017, the company entered into a collaboration and license agreement with BMS, under which BMS has the worldwide license to develop and commercialize products combining its ENHANZE Technology with BMS immuno-oncology targets directed at up to eleven targets for an upfront payment of $105.0 million payable on the effective date of the agreement. BMS has designated multiple immuno-oncology targets including programmed death 1 (PD-1) and has an option to select additional targets within five years from the effective date. The agreement became effective in November 2017 upon expiration of the waiting period provided by the Hart-Scott-Rodino Act.
- In September 2017, the company entered into an agreement with Roche for the right to develop and commercialize one additional exclusive target using its ENHANZE Technology for an upfront payment of $30.0 million.
- In August 2017, Lilly initiated a Phase 1 study of an investigational new therapy in combination with rHuPH20.
- In July 2017, Genentech initiated a Phase 1b/2 clinical trial evaluating PEGPH20 in combination with Tecentriq in patients with previously treated metastatic PDA.
Product and Product Candidates
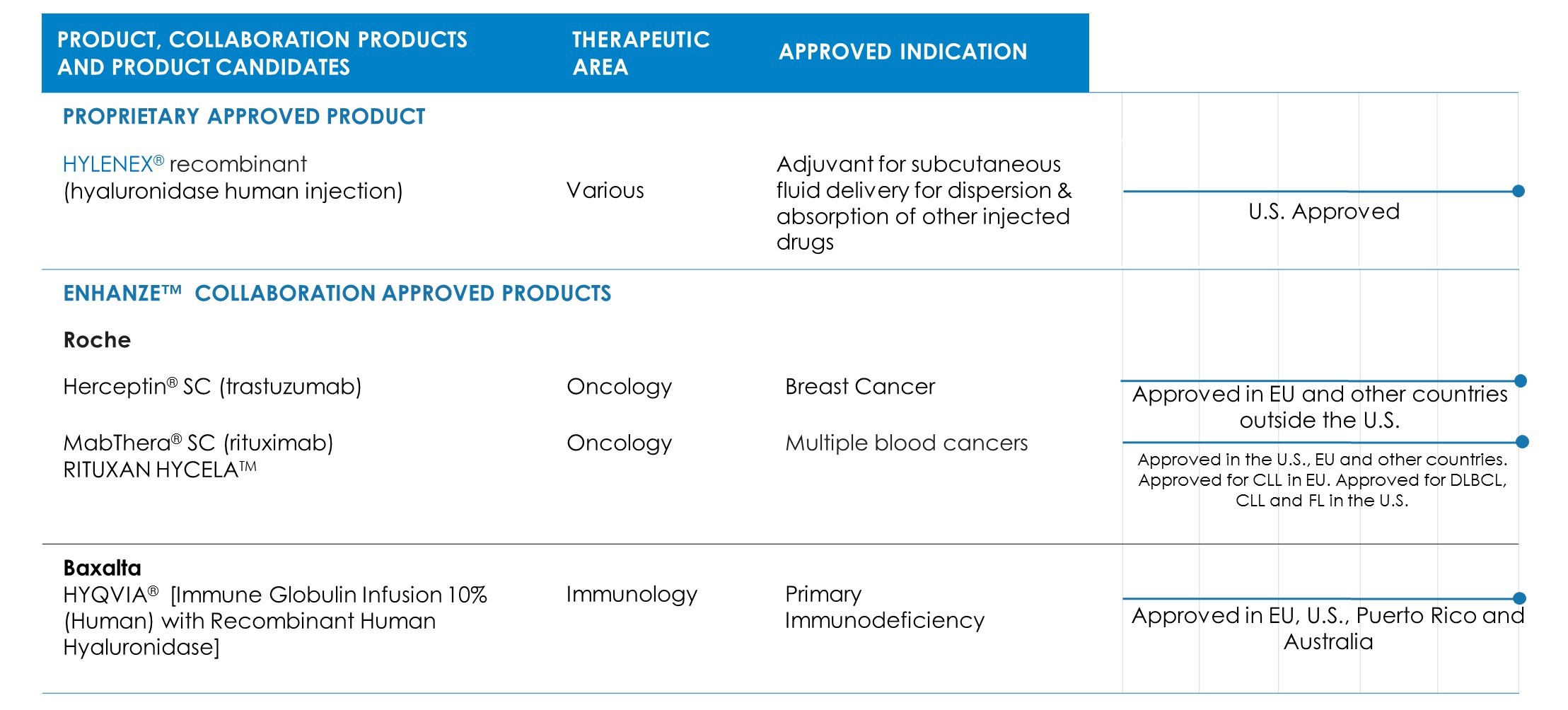
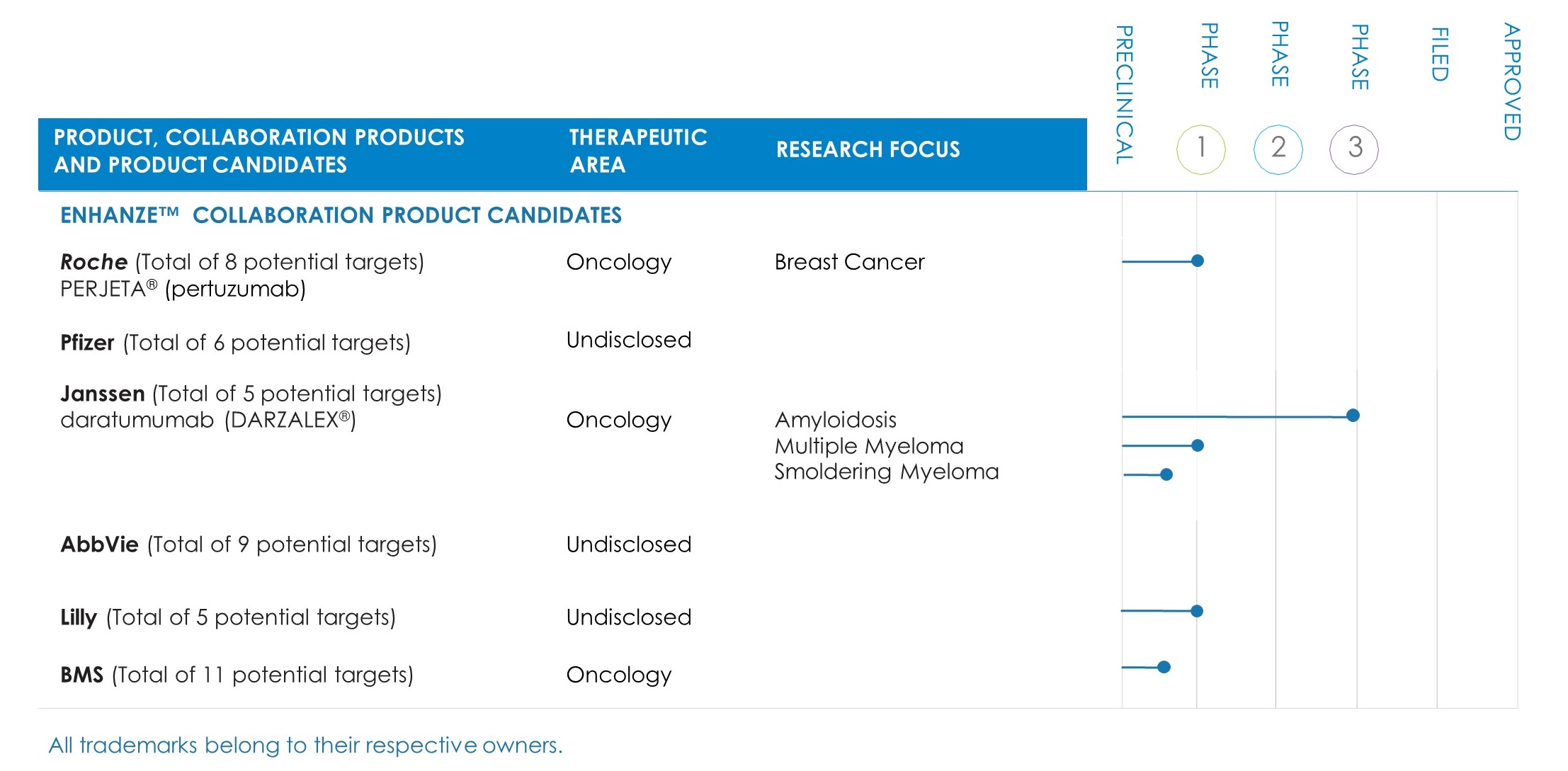
Halozyme Therapeutics has one marketed proprietary product, three partnered products, one proprietary product candidate targeting several indications in various stages of development, and two preclinical product candidates. The following table summarizes its proprietary product and product candidate as well as products and product candidates under development with its collaborators:
Proprietary Pipeline
Hylenex Recombinant (hyaluronidase human injection)
Hylenex recombinant is a formulation of rHuPH20 that has received U.S. Food and Drug Administration (FDA) approval to facilitate subcutaneous fluid administration for achieving hydration, to increase the dispersion and absorption of other injected drugs and, in subcutaneous urography, to improve resorption of radiopaque agents. Hylenex recombinant is currently the number one prescribed branded hyaluronidase.
PEGPH20
Halozyme Therapeutics is developing PEGPH20 in combination with currently approved cancer therapies as a candidate for the systemic treatment of tumors that accumulate HA. ‘PEG’ refers to the attachment of polyethylene glycol to rHuPH20, thereby creating PEGPH20. One of the novel properties of PEGPH20 is that it lasts for an extended duration in the bloodstream and, therefore, can be administered systemically to maintain its therapeutic effect to treat disease.
Cancer malignancies, including pancreatic, lung, breast, gastric, colon and prostate cancers can accumulate high levels of HA and therefore the company believe that PEGPH20 has the potential to help patients with these types of cancer when used with certain currently approved cancer therapies. Among solid tumors, PDA has been reported to be associated with the highest frequency of HA accumulation. There are approximately 65,000 annual diagnoses of PDA in the United States and the European Union, and the company estimate that 35-40% have high levels of HA.
The pathologic accumulation of HA, along with other matrix components, creates a unique microenvironment for the growth of tumor cells compared to normal cells. The company believe that degrading the HA component of the tumor microenvironment with PEGPH20 remodels the tumor microenvironment, resulting in tumor growth inhibition in animal models. Removal of HA from the tumor microenvironment results in expansion of previously constricted blood vessels allowing increased blood flow, potentially increasing the access of activated immune cells and factors in the blood into the tumor microenvironment. If PEGPH20 is administered in conjunction with other anti-cancer therapies, the increase in blood flow may allow anti-cancer therapies to have greater access to the tumor, which may enhance the treatment effect of therapeutic modalities like chemotherapies, monoclonal antibodies and other agents.
Halozyme Therapeutics is developing PEGPH20 as a targeted therapy, for patients who have tumors with high levels of HA. Halozyme Therapeutics has a collaboration with Ventana Medical Systems Inc. (Ventana), a member of the Roche Group, to develop, and for Ventana to ultimately commercialize, a companion diagnostic assay for use with PEGPH20. The companion diagnostic assay is being used to identify high levels of HA in tumor biopsies, and may be the first diagnostic to target tumor-associated HA and possibly the first companion diagnostic assay in pancreatic cancer.
Pancreatic cancer indications:
Halo 109-201:
In January 2015, the company presented the final results from Halo 109-201, a multi-center, international open label dose escalation Phase 1b clinical study of PEGPH20 in combination with gemcitabine for the treatment of patients with stage IV PDA at the 2015 Gastrointestinal Cancers Symposium (also known as ASCO-GI meeting). This study enrolled 28 patients with previously untreated stage IV PDA. Patients were treated with one of three doses of PEGPH20 (1.0, 1.6 and 3.0 µg/kg twice weekly for four weeks, then weekly thereafter) in combination with gemcitabine 1000 mg/m2 administered intravenously. In this study, the confirmed overall response rate (complete response + partial response confirmed on a second scan as assessed by an independent radiology review) was 29 percent (7 of 24 patients) for those treated at therapeutic dose levels of PEGPH20 (1.6 and 3.0 µg/kg). Median progression-free survival (PFS) was 154 days (95% CI, 50-166) in the efficacy-evaluable population (n = 24). Among efficacy-evaluable patients with baseline tumor HA staining (n = 17), the median PFS in patients with high baseline tumor HA staining (6/17 patients) was substantially longer, 219 days, than in the patients with low baseline tumor HA staining (11/17 patients), 108 days. Median overall survival (OS) was 200 days (95% CI, 123-370) in the efficacy-evaluable population (n = 24). Among efficacy-evaluable patients with baseline tumor HA staining (n = 17), the median OS in patients with high baseline tumor HA staining (6/17 patients) was substantially longer, 395 days, than in the patients with low baseline tumor HA staining (11/17 patients), 174 days. The most common treatment-emergent adverse events (occurring in ≥ 15% of patients) were peripheral edema, muscle spasms, thrombocytopenia, fatigue, myalgia, anemia, and nausea. Thromboembolic (TE) events were reported in 8 patients (28.6%) and musculoskeletal events were reported in 21 patients (75%) which were generally grade 1/2 in severity.
Halo 109-202:
In the second quarter of 2013, the company initiated Halo 109-202, a Phase 2 multicenter randomized clinical trial evaluating PEGPH20 as a first-line therapy for patients with stage IV PDA. The study was designed to enroll patients who would receive gemcitabine and nab-paclitaxel (ABRAXANE®) either with or without PEGPH20. The primary endpoint is to measure the improvement in PFS in patients receiving PEGPH20 plus gemcitabine and ABRAXANE (PAG arm) compared to those who are receiving gemcitabine and ABRAXANE alone (AG arm). In April 2014, after 146 patients had been enrolled, the trial was put on clinical hold by Halozyme and the FDA to assess a question raised by the Data Monitoring Committee regarding a possible difference in the TE events rate between the group of patients treated in the PAG arm versus the group of patients treated in the AG arm. This portion of the study and patients in this portion are now referred to as Stage 1. At the time of the clinical hold all patients remaining in the study continued on gemcitabine and ABRAXANE. In July 2014, Halo 109-202 was reinitiated (Stage 2) under a revised protocol, which excludes patients that are expected to be at a greater risk for TE events. The revised protocol provides for thromboembolism prophylaxis of all patients in both arms of the study with low molecular weight heparin, and adds evaluation of the TE events rate in Stage 2 PEGPH20-treated patients as a co-primary end point. Stage 2 of Halo 109-202 enrolled an additional 133 patients, to add to the 146 patients already in the clinical trial, with a 2:1 randomization for the PAG arm compared to the AG arm.
In March 2016, its partner Ventana received approval for an investigational device exemption (IDE) application from the FDA for its companion diagnostic test to enable patient selection in its Phase 3 Study 301 of PEGPH20 in HA-High patients. Based on the cutpoint for the Ventana diagnostic, the company expect approximately 35 to 40 percent of stage IV PDA patients to have HA-High tumors, similar to the previously reported interim results from Stage 1 of Study 202 using the Halozyme prototype assay.
In January 2017, the company announced topline results from the combined analysis of Stage 1 and Stage 2, and Stage 2 alone, based on a December 2016 data cutoff. The combined analysis included 135 treated patients in Stage 1, of whom a total of 45 patients (25 in the PAG arm and 21 in the AG arm) were determined to have high HA, and 125 treated patients in Stage 2, of whom a total of 35 patients (24 in the PAG arm and 11 in the AG arm) were determined to have high HA. This analysis of secondary and exploratory endpoints was conducted using the Ventana companion diagnostic to prospectively identify high levels of HA. The key results showed in the combined Stage 1 and Stage 2 dataset:
- The primary endpoint of PFS in the efficacy evaluable population (total of 231 patients) was met with statistical significance with a median PFS of 6.0 months in the PAG arm compared to 5.3 months in the AG arm, hazard ratio (HR) with a 95% confidence interval (CI): 0.73 (0.53, 1.00); p=0.048;
- The secondary endpoint of PFS in the HA-High intent to treat population (total of 84 HA-High patients) was met with statistical significance with a median PFS of 9.2 months in the PAG arm compared to 5.2 months in the AG arm, HR 0.51 (95% CI: 0.26, 1.00); p=0.048;
- The exploratory analysis of median OS was 11.5 months vs. 8.5 months in the PAG vs. AG arms, respectively. Factors potentially having an impact on these results include less aggressive disease among patients in the AG arm within the Stage 1 patient population, and 9 of the 24 patients in the PAG arm (approximately 40 percent) discontinued PEGPH20 treatment at the time of the clinical hold, resulting in many patients receiving AG alone in both arms.
- In the Stage 2 cohort population, in a total of 35 HA-High patients, the key results showed:
- Median PFS was 8.6 months in the PAG arm compared to 4.5 months in the AG arm, hazard ratio of 0.63 (95% CI: 0.21, 1.93);
- Median overall survival (OS) was 11.7 months in the PAG arm compared to 7.8 months in the AG arm, hazard ratio of 0.52 (95% CI: 0.22, 1.23);
The primary safety endpoint of decreasing the rate of TE events in Stage 2 was also met with the rate of TE events reducing from 43 percent to 10 percent in the PAG arm and from 25 percent to 6 percent in the AG arm, following a protocol amendment that excluded patients at high risk of TE events and with the introduction of prophylaxis with low molecular weight heparin (enoxaparin) in Stage 2 of the study with the current 1mg/kg/day dose of enoxaparin prophylaxis given in both treatment arms of the study.
In June 2017, results from Study 202 were presented at the ESMO World Congress of Gastrointestinal Cancer and the Annual Meeting of the American Society of Clinical Oncology (ASCO). Study 202 is an ongoing study with an open database and therefore the company continue to collect and receive data on both Stage 1 and Stage 2 patients. When the database is considered complete and locked, an updated analysis and Final Study Report will be generated.
Halo 109-301:
In March 2015, the company met with the FDA to discuss both the interim efficacy and safety data from Halo 109-202, which included the potential risk profile including TE event rate. Based on the feedback from that meeting, the company proceeded with a Phase 3 clinical study (Halo 109-301) of PEGPH20 in patients with stage IV PDA, using a design allowing for potential marketing application based on PFS (accelerated approval pathway) or OS. The study will enroll patients whose tumors accumulate high levels of HA measured using the Ventana companion diagnostic test. The FDA provided feedback on the current companion diagnostic approach and confirmed that an approved IDE is required for the Phase 3 study.
The use of PFS as the basis for marketing approval will be subject to the overall benefit and risk associated with PEGPH20 combined with gemcitabine and ABRAXANE therapy, including the:
- Magnitude of the PFS treatment effect observed;
- Toxicity profile; and
- Interim OS data.
In June 2015, the company received scientific advice/protocol assistance from the European Medicines Agency (EMA) regarding its Phase 3 study. The EMA agreed to the patient population, and the use of both PFS and OS as co-primary endpoints stating that OS is the preferred endpoint and that ultimate approval would require an overall positive benefit:risk balance.
In March 2016, the company dosed the first patient in Halo 109-301, a Phase 3 multicenter randomized clinical trial evaluating PEGPH20 as a first-line therapy for patients with stage IV PDA. The study will evaluate the effects on PFS and OS of PEGPH20 with gemcitabine and ABRAXANE compared with gemcitabine and ABRAXANE alone in stage IV PDA patients. In September 2017, its independent Data Safety Monitoring Committee met to review ongoing safety data from the trial and informed it the study should proceed as planned. Approximately 220 sites in 22 countries located in North America, Europe, South America and Asia have been initiated to participate in the HALO 301 study. An interim analysis will be conducted for its first primary endpoint when the company achieve the target number of PFS events. The company project that the target number of PFS events will be achieved in the fourth quarter of 2018. At that time the company project the company will have enrolled approximately 500 patients.
SWOG Study S1313:
In October 2013, SWOG, a cancer research cooperative group of more than 4,000 researchers in over 500 institutions around the world, initiated a 144 patient Phase 1b/2 randomized clinical trial in some of their study centers, examining PEGPH20 in combination with modified FOLFIRINOX chemotherapy compared to modified FOLFIRINOX treatment alone in patients with stage IV PDA (funded by the National Cancer Institute). As announced in March 2017, SWOG stopped enrollment in the Phase 1b/2 trial. While PEGPH20 is a targeted investigational therapy for patients with high levels of HA, the SWOG study was enrolling patients irrespective of HA levels, referred to as an all-comer population. During a planned early futility analysis, SWOG’s independent Data Monitoring Committee found, based on preliminary data, that the addition of PEGPH20 given every two weeks to modified FOLFIRINOX in this all-comer population would be unlikely to demonstrate a statistically significant improvement in the primary endpoint of overall survival. SWOG further reported that a higher rate of death was observed in the PEGPH20 arm versus modified FOLFIRINOX alone. SWOG has stopped the study and continues its ongoing effort to collect and clean outstanding data. Upon completion, the company will work with SWOG to analyze and evaluate the dataset. The company's PEGPH20 studies and clinical collaborations in combination with agents other than modified FOLFIRINOX continue unchanged.
Clinical collaboration:
In October 2016, the company announced that PEGPH20 will be included in a pancreatic cancer clinical trial initiative called Precision Promise, an initiative that aims to change the current treatment approach to pancreatic cancer by offering options to patients based on the molecular profile of their tumor. This is being accomplished through the Pancreatic Cancer Action Network leading a collaboration that brings together clinicians, researchers, and drug developers. Pancreatic Cancer Action Network continues to work to finalize the trial design and protocol which is expected to start in 2018.
Other indications outside of pancreatic cancer :
Halo 107-101:
In November 2015, the company initiated a Phase 1b study exploring the combination of PEGPH20 and KEYTRUDA®, an immuno-oncology agent in relapsed non-small cell lung cancer (NSCLC) and gastric cancer. In December 2016, the company identified a dose of PEGPH20, namely 2.2 ug/kg, to move into the dose expansion phase of the study with KEYTRUDA in combination with PEGPH20. Halozyme Therapeutics is enrolling both NSCLC and gastric cancer patients prospectively based on a patient being determined to be HA-High using the Ventana companion diagnostic test. In September 2017, its standing Independent Data Monitoring Safety Committee met to review ongoing safety data from the trial and informed it that the study should proceed with study protocol modifications to exclude patients at risk and increase liver safety monitoring, after observing clinical and laboratory signs of hepato-biliary dysfunction.
Clinical collaborations:
In July 2015, the company entered into a clinical collaboration agreement with Eisai Co., Ltd. (Eisai) to evaluate Eisai's HALAVEN® (eribulin) with PEGPH20 in HER2-negative metastatic breast cancer. In July 2016, the first patient was dosed in a Phase 1b/2 study for patients treated with up to two lines of prior therapy for HER2-negative HA-High metastatic breast cancer. Halozyme and Eisai are jointly sharing the costs to conduct this global study which continues in Phase 1b.
In November 2016, the company entered into an agreement with Genentech, a member of the Roche Group, to collaborate on clinical studies to evaluate their cancer immunotherapy Tecentriq, an anti-PD-L1 monoclonal antibody, in combination with PEGPH20, in up to eight different tumor types. Genentech initiated a Phase 1b/2 clinical trial in patients with previously treated metastatic PDA in July 2017 and a Phase 1b/2 clinical trial in patients with gastric cancer in October 2017, as part of its Morpheus master protocol. The company will supply PEGPH20 for the Genentech-funded studies. In October 2017, the company initiated a Phase 1b/2 clinical trial to assess Tecentriq with PEGPH20 in patients with cholangiocarcinoma and gall bladder cancer (Halo 110-101/MATRIX). Genentech will supply Tecentriq for the Halozyme sponsored study.
Regulatory
The FDA has granted Fast Track designation for its program investigating PEGPH20 in combination with gemcitabine and nab-paclitaxel for the treatment of patients with stage IV PDA to demonstrate an improvement in OS. The Fast Track designation process was developed by the FDA to facilitate the development and expedite the review of drugs to treat serious or life-threatening diseases and address unmet medical needs.
The FDA has granted Orphan Drug designation for PEGPH20 for the treatment of pancreatic cancer. The FDA Office of Orphan Products Development’s mission is to advance the evaluation and development of products (drugs, biologics, devices, or medical foods) that demonstrate promise for the diagnosis and/or treatment of rare diseases or conditions. Similarly, the European Committee for Orphan Medicinal Products of the EMA designated PEGPH20 an orphan medicinal product for the treatment of pancreatic cancer.
Other Pipeline Assets
HTI-1511: HTI-1511 is a novel antibody-drug conjugate (ADC) targeting epidermal growth factor receptor (EGFR) to treat solid tumors, including those with drug-resistant mutations. Halozyme Therapeutics is exploring potential collaboration or partnership interest in this program prior to making additional investments in the development of HTI-1511.
PEG-ADA2: PEGylated adenosine deaminase 2, or PEG-ADA2, is an immune checkpoint inhibitor that targets adenosine, which may accumulate to high levels in the tumor microenvironment and has been linked to immunosuppression. Halozyme Therapeutics is currently in preclinical development with PEG-ADA2 and are exploring potential collaboration or partnership interest in this program prior to making additional investments in the development of PEG-ADA2.
ENHANZE Collaborations
Roche Collaboration
In December 2006, the company and Roche entered into a collaboration and license agreement under which Roche obtained a worldwide, license to develop and commercialize product combinations of rHuPH20 and up to thirteen Roche target compounds (the Roche Collaboration). Roche initially had the exclusive right to apply rHuPH20 to three pre-defined Roche biologic targets with the option to develop and commercialize rHuPH20 with ten additional targets. Roche had the right to exercise this option to identify additional targets for ten years. As of the ten year anniversary of the Roche Collaboration in December 2016, Roche had elected a total of eight targets, two of which are exclusive.
In September 2013, Roche launched a subcutaneous (SC) formulation of Herceptin (trastuzumab) (Herceptin SC) in Europe for the treatment of patients with HER2-positive breast cancer. This formulation utilizes its patented ENHANZE Technology and is administered in two to five minutes, compared to 30 to 90 minutes with the standard intravenous form. Roche received European marketing approval for Herceptin SC in August 2013. Breast cancer is the most common cancer among women worldwide. HER2-positive cancer is reported to be a particularly aggressive form of breast cancer. Directed at the same target, Roche initiated a Phase 1 study of rHuPH20 with PERJETA® (pertuzumab) in patients with early breast cancer in March 2016.
In June 2014, Roche launched MabThera SC in Europe for the treatment of patients with common forms of non-Hodgkin lymphoma (NHL). This formulation utilizes its patented ENHANZE Technology and is administered in approximately five minutes compared to the approximately 1.5 to 4 hour infusion time for intravenous MabThera. The European Commission approved MabThera SC in March 2014. In May 2016, Roche announced that the EMA approved Mabthera SC to treat patients with chronic lymphocytic leukemia (CLL). In June 2017, the FDA approved Genentech’s (a member of the Roche Group) RITUXAN HYCELA™, a combination of rituximab and rHuPH20 (approved and marketed under the MabThera SC brand in countries outside the U.S.), for CLL and two types of NHL, follicular lymphoma and diffuse large B-cell lymphoma.
In September 2017, the company and Roche entered into an agreement providing Roche the right to develop and commercialize one additional exclusive target using its ENHANZE Technology (the 2017 Roche Collaboration). The upfront license payment may be followed by event-based payments subject to Roche’s achievement of specified development, regulatory and sales-based milestones. In addition, Roche will pay royalties to it if products under the collaboration are commercialized.
Baxalta Collaboration
In September 2007, the company and Baxalta entered into a collaboration and license agreement under which Baxalta obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20 with GAMMAGARD LIQUID (HYQVIA) (the Baxalta Collaboration). HYQVIA is indicated for the treatment of primary immunodeficiency disorders associated with defects in the immune system.
In May 2013, the European Commission granted Baxalta marketing authorization in all EU Member States for the use of HYQVIA (solution for subcutaneous use) as replacement therapy for adult patients with primary and secondary immunodeficiencies. Baxalta launched HYQVIA in the first EU country in July 2013 and has continued to launch in additional countries.
In September 2014, HYQVIA was approved by the FDA for treatment of adult patients with primary immunodeficiency in the U.S. HYQVIA is the first subcutaneous immune globulin (IG) treatment approved for adult primary immunodeficiency patients with a dosing regimen requiring only one infusion up to once per month (every three to four weeks) and one injection site per infusion in most patients, to deliver a full therapeutic dose of IG. Prior to the approval of HYQVIA, the majority of primary immunodeficiency patients received intravenous infusions in a doctor’s office or infusion center, and other subcutaneous IG treatments require weekly or bi-weekly treatment with multiple infusion sites per treatment. The FDA’s approval of HYQVIA was a significant milestone for it as it represented the first U.S. approved BLA which utilizes its rHuPH20 platform.
In May 2016, Baxalta announced that HYQVIA received a marketing authorization from the European Commission for a pediatric indication, which is being launched in Europe to treat primary and certain secondary immunodeficiencies.
Pfizer Collaboration
In December 2012, the company and Pfizer entered into a collaboration and license agreement, under which Pfizer has the worldwide license to develop and commercialize products combining its rHuPH20 enzyme with Pfizer proprietary biologics directed to up to six targets in primary care and specialty care indications. Targets may be selected on an exclusive or non-exclusive basis. Pfizer has elected five targets on an exclusive basis and returned 2 targets.
Janssen Collaboration
In December 2014, the company and Janssen entered into a collaboration and license agreement, under which Janssen has the worldwide license to develop and commercialize products combining its rHuPH20 enzyme with Janssen proprietary biologics directed to up to five targets. Targets may be selected on an exclusive basis. Janssen has elected CD38 as the first target on an exclusive basis. In November 2015, Janssen initiated dosing in a Phase 1b clinical trial evaluating subcutaneous delivery of daratumumab, directed at CD38, using ENHANZE Technology, in multiple myeloma patients. In December 2016, Janssen announced results of the trial, which supported continued development of daratumumab with rHuPH20. In October 2017, Janssen initiated a Phase 3 study of daratumumab combined with the ENHANZE Technology in amyloidosis patients, with additional Phase 3 studies in multiple myeloma and smoldering myeloma patients planned to initiate in the near term.
AbbVie Collaboration
In June 2015, the company and AbbVie entered into a collaboration and license agreement, under which AbbVie has the worldwide license to develop and commercialize products combining its rHuPH20 enzyme with AbbVie proprietary biologics directed to up to nine targets. Targets may be selected on an exclusive basis. AbbVie elected one target on an exclusive basis, TNF alpha, for which it has discontinued development and returned the target.
Lilly Collaboration
In December 2015, the company and Lilly entered into a collaboration and license agreement, under which Lilly has the worldwide license to develop and commercialize products combining its rHuPH20 enzyme with Lilly proprietary biologics directed to up to five targets. Targets may be selected on an exclusive basis. Lilly has elected two targets on an exclusive basis and one target on a semi-exclusive basis. In August 2017, Lilly initiated a Phase 1 study of an investigational new therapy in combination with rHuPH20.
BMS Collaboration
In September 2017, the company and BMS entered into a collaboration and license agreement, under which BMS will have the worldwide license to develop and commercialize products combining its rHuPH20 enzyme with BMS immuno-oncology targets directed at up to eleven targets upon effectiveness of the agreement. The agreement became effective in November 2017 upon expiration of the waiting period provided by the Hart-Scott-Rodino Act. BMS has designated multiple immuno-oncology targets including programmed death 1 (PD-1) and has an option to select additional targets within five years from the effective date.




