Intercept Pharmaceuticals
Overview
Intercept Pharmaceuticals (ICPT) is a biopharmaceutical company focused on the development and commercialization of novel therapeutics to treat non-viral, progressive liver diseases with high unmet medical need utilizing its proprietary bile acid chemistry. The company's one marketed product, OCA, and portfolio of clinical product candidates have the potential to treat orphan and more prevalent liver diseases for which, currently, there are limited therapeutic solutions.1
OCA was approved in the United States in May 2016 for use in patients with PBC, under the brand name Ocaliva® (obeticholic acid). OCA is a bile acid analog, a chemical substance that has a structure based on a naturally occurring human bile acid, that selectively binds to and activates the farnesoid X receptor, or FXR. The company believe OCA has broad liver-protective properties and may effectively counter a variety of chronic insults to the liver that cause fibrosis, or scarring, which can eventually lead to cirrhosis, liver transplant and death. The company commenced sales and marketing of Ocaliva in the United States shortly after receiving such marketing approval, and Ocaliva is now available to patients primarily through a network of specialty pharmacy distributors. In December 2016, the European Commission granted conditional approval for Ocaliva for the treatment of PBC and the company commenced its European commercial launch in January 2017. Intercept Pharmaceuticals has submitted or are in the process of submitting dossiers to a number of reimbursement authorities in the European Union. In May 2017, Health Canada granted a conditional approval for Ocaliva in PBC and the company commenced its commercial launch in July 2017. The company also plan to file for marketing authorization for OCA in PBC in other target markets.
Intercept Pharmaceuticals is currently evaluating its future development strategy for OCA in other indications, including a variety of other non-viral progressive liver diseases such as nonalcoholic steatohepatitis, or NASH, primary sclerosing cholangitis, or PSC, and biliary atresia.
OCA achieved the primary endpoint in a Phase 2b clinical trial for the treatment of NASH, known as the FLINT trial, which was sponsored by the U.S. National Institute of Diabetes and Digestive and Kidney Diseases, or NIDDK, a part of the National Institutes of Health. The FLINT trial was completed in late July 2014. Intercept Pharmaceuticals has an ongoing Phase 3 clinical trial in non-cirrhotic NASH patients with liver fibrosis, known as the REGENERATE trial. REGENERATE includes a pre-planned histology-based interim analysis after 72 weeks of treatment. In May 2017, the company completed enrollment of the interim analysis cohort for the REGENERATE trial. The company anticipate top-line results from the interim analysis in the first half of 2019. Intercept Pharmaceuticals has also completed a Phase 2 clinical trial, known as the CONTROL trial, the goal of which was to characterize the lipid metabolic effects of OCA and cholesterol management effects of concomitant statin administration in NASH patients. The company announced that this trial met its primary endpoint in July 2017. The company continue to work towards expanding its overall NASH development program with additional trials and studies, including its ongoing Phase 3 trial in NASH patients with compensated cirrhosis, known as the REVERSE trial, which the company announced in February 2018.
In addition to PBC and NASH, the company continue to invest in research of OCA for additional patient populations with other liver diseases. For example, in July 2017, the company announced top-line results of its Phase 2 AESOP trial in PSC which evaluated the effects of 24 weeks of treatment with varying doses of OCA compared to placebo. This trial achieved its primary endpoint, which the company believe establishes a proof-of-concept of OCA in a second cholestatic liver disease. The company plan to discuss these results with regulatory authorities to formulate its future development plans for OCA in PSC.OCA has received orphan drug designation in the United States and the European Union for the treatment of PBC and PSC and breakthrough therapy designation from the U.S. Food and Drug Administration, or FDA, for the treatment of NASH patients with liver fibrosis.
Production Pipeline
OCA Development on Liver Diseases with Limited/No Approved Therapies The following chart shows the current stage of development of OCA in different patient populations:
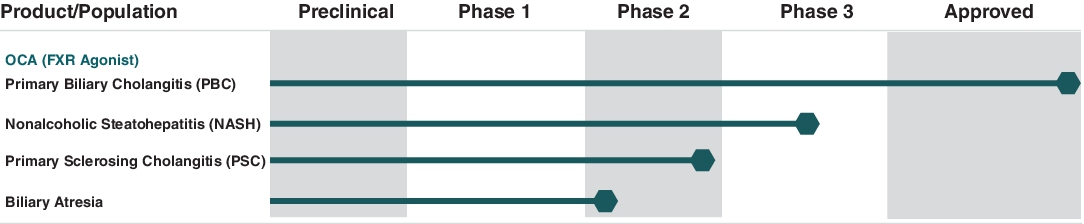
The company's current patents for OCA are scheduled to expire at various times through 2033. The company own or have rights to various trademarks, copyrights and trade names used in its business, including rights to OCA worldwide except for China, where Intercept Pharmaceuticals has exclusively licensed OCA to Sumitomo Dainippon Pharma Co. Ltd., or Sumitomo Dainippon.
By virtue of its patent portfolio and the proprietary know-how of its employees and its collaborators at the University of Perugia, the company believe that the company hold a leading position in the fields of bile acid chemistry and therapeutics. Starting with OCA and its underlying patents, which were assigned to it under its agreements with Professor Roberto Pellicciari, Ph.D., one of its co-founders, other researchers and the University of Perugia, its collaboration has resulted in a pipeline of bile acid analogs in addition to OCA. Through its collaboration with Professor Pellicciari and TES Pharma Srl, Intercept Pharmaceuticals is continuing its research to rationally design compounds that bind selectively and potently to FXR and other bile acid receptors.
Ocaliva Label Update
In the course of its post-marketing pharmacovigilance activities, deaths have been reported in PBC patients prescribed OCA with moderate or severe hepatic impairment. In an analysis performed by it and in consultation with the FDA, the company concluded that these patients were prescribed once daily doses of Ocaliva, which is seven times higher than the recommended weekly dose in such patients. As a result, in September 2017, the company issued a dear healthcare provider letter and the FDA also subsequently issued its own safety communication to reinforce recommended dosing in accordance with the Ocaliva label. Both communications reminded healthcare providers of the importance of the recommended reduced dosing of Ocaliva in PBC patients with moderate or severe hepatic impairment, while reiterating the importance of monitoring PBC patients for progression of their disease and the occurrence of liver-related adverse reactions.
In February 2018, the company announced that the Ocaliva label in the United States had been updated by the FDA to include a boxed warning and a dosing table that reinforce the existing dosing schedule for patients with Child-Pugh Class B, or C or decompensated cirrhosis. In addition, the FDA issued an updated drug safety communication to accompany the revised label. The company remain focused on the safety of all of the patients using Ocaliva within and outside of its ongoing clinical studies and are working with relevant regulatory authorities, including the European Medicines Agency, or EMA, to ensure that the Ocaliva label in such jurisdictions sufficiently reinforces the importance of appropriate dosing in patients with advanced cirrhosis.
Strategy
The company's strategy is to develop and commercialize novel therapeutics for patients with progressive non-viral liver diseases, beginning with OCA for the treatment of PBC, NASH and other follow-on indications that the company believe are underserved by existing marketed therapies. The key elements of its strategy are to:
- commercialize OCA in the United States, Europe, Canada and other countries, initially for the treatment of PBC;
- continue to develop OCA for the treatment of NASH and seek regulatory approval of OCA in this indication;
- continue to develop OCA in other orphan and more prevalent liver diseases; and
- maintain infrastructure and personnel in the United States and internationally to support its product development and commercialization efforts, as well as its operations as a public company.
In order to achieve its strategic objectives, Intercept Pharmaceuticals has, and will remain, focused on hiring and retaining a highly skilled management team and employee base with extensive experience and specific skill sets relating to the selection, development and commercialization of therapies for liver diseases with high unmet medical need.
Overview of Liver Function, Bile Acids and Chronic Liver Diseases The liver performs many functions that are vital for maintaining health, including the regulation of bile acid metabolism. Bile acids are natural detergent-like emulsifying agents that are released from the gallbladder into the intestine when food is ingested, and are essential for the absorption of dietary cholesterol and other nutrients. Cholesterol bound by bile acids is taken up by the liver, where the cholesterol is then converted into one of two primary bile acids. The bile acids are then actively secreted into bile ducts, which eventually empty into the gallbladder. This digestive cycle of bile flow from gallbladder to intestine to liver and back is called the enterohepatic recirculation of bile.
In addition to facilitating nutrient absorption, bile acids act as important signals that help regulate multiple other biological functions. They are also complex signaling molecules that integrate metabolic and immune pathways involved in the healthy functioning of various tissues and organs. For example, the actions of bile acids in the liver, intestine and kidney regulate repair mechanisms that modulate inflammation and fibrosis, or scarring, which can lead to progressive organ damage.
The biological effects of bile acids are mediated through dedicated receptors. The best understood receptor is FXR, a nuclear receptor that regulates bile acid synthesis and clearance from the liver, thereby preventing excessive bile acid build-up in the liver, which may be toxic. As such, FXR is a target for the treatment of several liver diseases such as PBC and PSC that involve impaired bile flow, a condition called cholestasis. In cholestasis, the liver is typically exposed to higher than normal levels of bile acids, which can cause significant damage over time. In addition, bile acid activation of FXR induces anti-fibrotic, anti-inflammatory, anti-steatotic and other mechanisms that are necessary for the normal regeneration of the liver. As a result, FXR is also a target for the treatment of more common liver diseases such as NASH and alcoholic hepatitis. Further, based on the discovery of similar FXR-mediated protective mechanisms in other organs exposed to bile acids, the company believe that FXR may also be a potential target for the treatment of a number of intestinal, kidney and other diseases.
Ocaliva
Overview
OCA was approved in the United States in May 2016 under the accelerated approval pathway for use in patients with PBC under the brand name Ocaliva. The company commenced sales and marketing of Ocaliva in the United States shortly after receiving such marketing approval, and Ocaliva is now available to patients primarily through a network of specialty pharmacy distributors. In December 2016, the European Commission granted conditional approval for Ocaliva for the treatment of PBC and the company commenced its European commercial launch in certain markets in January 2017. Intercept Pharmaceuticals has submitted or are in the process of submitting dossiers to a number of reimbursement authorities in the European Union. In May 2017, Health Canada granted a conditional approval for Ocaliva in PBC and the company commenced its commercial launch in Canada in July 2017.
Primary Biliary Cholangitis (PBC)
PBC is a rare liver disease that primarily results from autoimmune destruction of the bile ducts that transport bile acids out of the liver, resulting in cholestasis. The build-up of bile acids in the liver damage liver cells. These damaged liver cells, in turn, release abnormal amounts of serum alkaline phosphatase, or ALP, a liver enzyme that is a key biomarker of the disease pathology. As shown in numerous clinical trials of treatment with ursodeoxycholic acid, available generically as ursodiol, a positive therapeutic response is primarily determined by sustained reduction of ALP levels, along with maintenance of normal bilirubin levels, indicating adequately compensated liver function. This biochemical improvement has been shown to correlate well with improved clinical outcomes such as transplant-free survival. As the disease progresses, it causes progressive liver damage marked by chronic inflammation and fibrosis. Despite its rarity, PBC is the most common cholestatic liver disease and is the second leading indication for liver transplant among women in the United States. Disease progression in PBC varies significantly, with median survival in untreated patients of 7.5 years if symptomatic at diagnosis and up to 16 years if asymptomatic at diagnosis. PBC patients whose disease is progressing have persistently elevated levels of ALP and other liver enzymes, with abnormal bilirubin levels heralding more advanced disease. Data from published long-term studies demonstrate that a significant portion of such patients with advancing disease progress to liver failure, transplant or death within five to ten years.
An estimated 90% of PBC patients are women, with approximately one in 1,000 women over the age of 40 afflicted by the disease. The mean age of diagnosis is about 40 years and the typical initial presentation occurs between the ages of 30 and 65 years. A majority of PBC patients are asymptomatic at the time of initial diagnosis, but most develop symptoms over time. Fatigue and pruritus, or itching, are the most common symptoms in PBC patients. Less common symptoms include dry eyes and mouth, as well as jaundice, which can be seen in more advanced disease. Based on the guidelines of the American Association for the Study of Liver Disease and the European Association for the Study of the Liver, the clinical diagnosis of PBC is established based on the presence of ![]() a positive anti-mitochondrial antibody, or AMA, a marker of this autoimmune disease seen in up to 95% of PBC patients, and (ii) elevated serum levels of ALP. In the earlier stages of PBC, ALP is often the only abnormally elevated liver enzyme, rising to between two to ten times higher than normal values. Bilirubin is a marker of liver function and is also monitored in PBC to provide an indication of how well the liver is functioning. Liver biopsy can be used to confirm the diagnosis of PBC, but is not required and is becoming less-frequently performed.
a positive anti-mitochondrial antibody, or AMA, a marker of this autoimmune disease seen in up to 95% of PBC patients, and (ii) elevated serum levels of ALP. In the earlier stages of PBC, ALP is often the only abnormally elevated liver enzyme, rising to between two to ten times higher than normal values. Bilirubin is a marker of liver function and is also monitored in PBC to provide an indication of how well the liver is functioning. Liver biopsy can be used to confirm the diagnosis of PBC, but is not required and is becoming less-frequently performed.
A number of published clinical studies have demonstrated that lower levels of ALP, both independently or in conjunction with normal bilirubin levels, correlate with a significant reduction in adverse clinical outcomes such as liver transplant and/or death in PBC patients. These studies include the result of meta-analyses of PBC clinical outcomes data of more than 6,000 PBC patients from 15 academic centers in eight countries that have been compiled by the Global PBC Study Group, which the company sponsored, as well as a dataset of over 6,000 PBC patients across the United Kingdom compiled by the UK PBC Group.
Ocaliva Phase 3 Clinical Trial in PBC
The approval of Ocaliva in the United States, Europe and Canada was supported by the results of the pivotal Phase 3 POISE trial, which was completed in March 2014. The POISE data showed that Ocaliva, at both a once-daily 10 mg dose and a once-daily 5 mg dose titrated to 10 mg, met the trial’s primary endpoint of achieving a reduction in ALP to below a threshold of 1.67 times the upper limit of normal, or ULN, with a minimum of a 15% reduction in ALP level from baseline, and a normal bilirubin level after 12 months of therapy. The percentage of patients meeting the POISE trial primary endpoint was 10% in the placebo group, 47% in the 10 mg Ocaliva group and 46% in the Ocaliva titration group (both dose groups p < 0.0001 as compared to placebo) in an intent-to-treat analysis. The placebo group experienced a mean decrease in ALP from baseline of 5%, compared to a significant mean decrease of 39% in the 10 mg Ocaliva dose group and 33% in the Ocaliva titration group (both dose groups p < 0.0001 as compared to placebo). Pruritus, generally mild to moderate, was the most frequently reported adverse event associated with Ocaliva treatment.
Ongoing Confirmatory Clinical Outcomes Trial and Other Post Marketing Requirements In connection with Ocaliva’s accelerated approval in the United States and conditional approval in the European Union, the company committed to conduct a Phase 4 confirmatory outcomes trial of Ocaliva, known as the COBALT trial, to support post-marketing regulatory requirements. The goal of the trial is to confirm that reduction of ALP with OCA treatment is associated with a longer-term benefit on liver-related clinical outcomes. This trial is currently enrolling patients and is expected to be completed on a post-marketing basis.
COBALT is designed to assess the effect of a once-daily dose of 5 mg or 10 mg of Ocaliva in approximately 430 PBC patients with an inadequate therapeutic response to ursodiol or who are unable to tolerate ursodiol. In this trial, eligible patients with PBC continue their ursodiol treatment, except for those patients unable to tolerate ursodiol, and are being randomized into one of two treatment arms of approximately 215 patients each. Patients are randomized to receive either placebo or Ocaliva starting at 5 mg and increasing over the course of the trial to 10 mg of Ocaliva based on tolerability. Dosing frequency will be determined by disease stage. The primary endpoint of the trial is based on clinical outcomes as measured by time to first occurrence of any of the following adjudicated events: death (all-cause), liver transplant, Model of End Stage Liver Disease, or MELD, score greater than 15, hospitalization due to variceal bleeding, encephalopathy or spontaneous bacterial peritonitis, uncontrolled ascites or hepatocellular carcinoma. The study evaluates subjects across the spectrum of PBC disease, including early and advanced PBC.
Intercept Pharmaceuticals has agreed to evaluate the safety and efficacy of Ocaliva in patients with moderate to severe hepatic impairment and as monotherapy in patients with PBC. Finally, Intercept Pharmaceuticals has also agreed to develop and characterize a lower dose formulation of Ocaliva to allow for once daily dosing in patients with moderate or severe hepatic impairment. Full approval for Ocaliva in PBC may be contingent upon the verification and description of clinical benefit in confirmatory trials.
In the course of its post-marketing pharmacovigilance activities, deaths have been reported in PBC patients with moderate or severe hepatic impairment. In an analysis performed by it and in consultation with the FDA, the company concluded that these patients were prescribed once daily doses of Ocaliva, which is seven times higher than the recommended weekly dose in such patients. As a result, in September 2017, the company issued a dear healthcare provider letter, or DHCP letter, and the FDA also subsequently issued its own drug safety communication to reinforce recommended label dosing. Both communications remind healthcare providers of the importance of the recommended reduced dosing of Ocaliva in PBC patients with moderate or severe hepatic impairment, while reiterating the importance of monitoring PBC patients for progression of their disease and the occurrence of liver-related adverse reactions. In addition to the DHCP letter, Intercept Pharmaceuticals has taken actions to enhance education about appropriate use of Ocaliva. These initiatives include: reeducating physicians on the label, with a focus on ensuring appropriate dosing for patients with moderate or severe hepatic impairment; enhancing monitoring of patients for liver-related adverse reactions; and completing adjudication of all reported cases of serious liver injury, including in patients with no or mild hepatic impairment.
In February 2018, the company announced that the Ocaliva label in the United States had been updated by the FDA to include a boxed warning and a dosing table that reinforce the existing dosing schedule for patients with Child-Pugh Class B, or C or decompensated cirrhosis. In addition, the FDA issued an updated drug safety communication to accompany the revised label. The company remain focused on the safety of all of the patients using Ocaliva within and outside of its ongoing clinical studies and are working with relevant regulatory authorities, including the European Medicines Agency, or EMA, to ensure that the Ocaliva label in such jurisdictions sufficiently reinforces the importance of appropriate dosing in patients with advanced cirrhosis.
PBC Market Opportunity
Prior to Ocaliva, the only approved drug for the treatment of PBC was ursodeoxycholic acid, available generically as ursodiol, which is widely considered the standard first line therapy for PBC patients. In patients for whom ursodiol is effective, the treatment slows the progression of PBC, reducing the likelihood of liver failure and the need for transplant.
According to its analysis of 2016 industry data, there are approximately 290,000 people with PBC in its target markets, consisting of the United States, certain European countries, Canada, Australia and New Zealand. Based on its analysis of this 2016 data, the company believe approximately 119,000 patients in its target markets have been diagnosed and are under the care of a physician for PBC. Intercept Pharmaceuticals is focusing its commercial efforts on the estimated 37,000 diagnosed PBC patients who have elevated ALP levels of at least 1.67 times ULN, despite receiving treatment with ursodiol. Of those PBC patients, approximately 15,000 are estimated to be in the United States and 22,000 in its target countries outside of the United States. In addition, the company believe another 8,000 patients in its target countries, including approximately 4,000 patients in the United States, are intolerant to ursodiol or have discontinued ursodiol treatment due to lack of efficacy. Finally, the company believe there are approximately an additional 35,000 patients in its target countries, including approximately 15,000 in the United States, who have an elevated ALP greater than ULN but less than 1.67 times ULN who may be treated with Ocaliva.
The company's estimates of the potential market opportunity for OCA for the treatment of PBC include a number of key assumptions related to prevalence rates, patients’ access to healthcare, diagnosis rates and patients’ response to or tolerance of OCA, which are based on available literature and epidemiology research in PBC, its industry knowledge gained through market research and other methods, industry publications, third-party research reports and other surveys.
Clinical Development Status
As part of its lifecycle management strategy for OCA, Intercept Pharmaceuticals is pursuing the clinical development of Ocaliva as a potential treatment for NASH, PSC and biliary atresia.
Nonalcoholic Steatohepatitis (NASH)
Overview of NASH
NASH is a common and progressive chronic liver disease caused by excessive fat accumulation in the liver, or steatosis, that induces inflammation and may lead to progressive fibrosis and cirrhosis, which may eventually result in liver failure and death. In NASH patients, for reasons that are not yet completely understood, steatosis and other factors such as insulin resistance induce chronic inflammation in the liver and may lead to progressive fibrosis and cirrhosis, followed by eventual liver failure and death. More than 20% of patients with NASH progress to cirrhosis within a decade of diagnosis and, compared to the general population, have a ten-fold greater risk of liver-related mortality. Owing to the rapidly increasing prevalence of the disease, NASH has become the second most common reason for liver transplant in the United States and is projected to become the leading indication for transplant in the next few years, overtaking both chronic hepatitis C infection and alcoholic liver disease. Additionally, NASH is now considered to be the leading, and a rapidly increasing, cause of hepatocellular carcinoma, or primary liver cancer, of which up to 40% of cases in NASH patients develop prior to developing cirrhosis.
Although difficult to precisely estimate, current epidemiology research estimates that the global prevalence of NASH is approximately 3 – 5% and is expected to increase markedly by 2030. Fibrosis is the most robust predictor of long-term overall mortality, liver transplantation, and liver-related events in patients with NASH. More than 30% of those patients are believed to have fibrosis of stage 2 or greater. Although the prevalence of NASH is lower in children, it has also become a serious disease burden in the pediatric population. Other common co-existing conditions such as obesity and type 2 diabetes, which are present in a majority of NASH patients, raise important risks. NASH has been linked in both developed and developing countries to the adoption of a Western diet, with increased consumption of processed foods containing polyunsaturated fatty acids and fructose.
Currently, a definitive diagnosis of NASH is based on a histologic assessment of a liver biopsy for several key features associated with NASH, including, but not limited to, steatosis, lobular inflammation and hepatocyte ballooning. However, several imaging and circulating biomarkers are being investigated as non-invasive diagnostic methods, including transient elastography (an ultrasound technology approved in Europe and more recently in the United States for the measurement of liver fibrosis), magnetic resonance imaging and serum biomarkers. NASH diagnosis rates in the United States and the EU5 countries are very low, owing to a lack of approved treatment options and a lack of validated non-invasive diagnosis options. The company believe the availability of novel therapeutics and non-invasive technologies will be instrumental in improving diagnosis rates.
Currently Available Treatment Options for NASH
There are currently no drugs approved for the treatment of NASH. However, various therapeutics are used off-label, such as vitamin E (an antioxidant), insulin sensitizers (e.g., metformin, pioglitazone), antihyperlipidemic agents (e.g., gemfibrozil), pentoxifylline and ursodiol. Lifestyle changes, including modification of diet and exercise to reduce body weight, as well as treatment of concomitant diabetes and dyslipidemia, are commonly accepted as the standard of care, but have not conclusively been shown to prevent disease progression.
NASH Unmet Medical Need
Although some of the off-label treatments described above have been studied as possible treatments for NASH, none has been approved by the FDA or EMA as a treatment for this disease. Currently, treatment options for NASH patients with advanced cirrhosis are limited. Although liver transplant can be life-saving, many patients fail to receive a donor organ in time, and for those who do, there are very significant clinical risks, such as infection and organ rejection, as well as significant costs. In addition, the post-transplant recurrence rate of NASH has been shown to be as high as 25% at 18 months. Given the lack of available treatment options, the company believe that there is a significant unmet need for novel therapies for NASH, particularly in those patients with advanced fibrosis and cirrhosis and those with a high risk of disease progression due to other co-morbidities such as type 2 diabetes.
Breakthrough Therapy Designation
In January 2015, OCA received breakthrough therapy designation from the FDA for the treatment of NASH patients with liver fibrosis. The breakthrough therapy designation was created by the FDA to speed the availability of new therapies for serious or life-threatening conditions. Drugs qualifying for this designation must show credible evidence of a substantial improvement on a clinically significant endpoint over available therapies, or over placebo if there is no available therapy. The breakthrough therapy designation constitutes one of four expedited programs for serious conditions including accelerated approval, priority review and fast-track designation, all of which can also be granted to the same drug if relevant criteria are met. The breakthrough therapy designation confers several benefits, including intensive FDA guidance and discussion and eligibility for submission of a rolling new drug application, or NDA.
Current Status of Clinical Trials
Intercept Pharmaceuticals has conducted or are conducting various clinical trials for the treatment of NASH, including clinical trials required in order to successfully obtain an NDA.
FXR activation has been shown to play a key role in the regulation of the metabolic pathways relevant to NASH, highlighting FXR as a potential drug target for treatment of the disease. Given the significant unmet medical need of patients with NASH, the company believe that the ability of OCA to potently activate FXR has the potential to convey clinical benefit through potential amelioration or reversal of liver fibrosis, inflammation, steatosis, and insulin resistance. This is supported by preclinical and clinical results obtained to date, and further investigated in its ongoing clinical trial program. For example, in animal models, sustained FXR activation with OCA treatment has resulted in the reversal of liver fibrosis, the reversal of portal hypertension, the prevention of atherosclerosis, and improvements in triglycerides, inflammation, steatosis and insulin sensitivity. Mice that lack functional FXR (so-called knockout mice) spontaneously develop NASH accompanied by hypertriglyceridemia and insulin resistance, and go on to develop hepatocellular carcinoma, or primary liver cancer. The company believe that the combined mechanisms of FXR activation, coupled with the occurrence of NASH in animals lacking FXR, support the potential disease-modifying therapeutic potential of OCA in directly addressing the underlying disease pathology in NASH.
(a) Phase 2 Trial in Type 2 Diabetic Patients with NAFLD
The company previously completed a double-blind, placebo-controlled Phase 2 clinical trial of OCA in 64 type 2 diabetic patients with NAFLD. In this trial, OCA therapy significantly improved insulin sensitivity both in the liver and peripheral tissues, thereby meeting the primary endpoint in the trial with a mean improvement in liver insulin sensitization from baseline of approximately 24.5% in the combined OCA dose groups, as compared to a worsening of approximately 5.5% in the placebo group (p = 0.011). Insulin resistance, particularly in the liver, is considered to be an important contributor to NASH disease pathology. In this trial, significant reductions in body weight were also noted in patients receiving OCA therapy, along with improvements in liver enzymes such as gamma-glutamyl transferase, or GGT, and aspartate transaminase, or AST.
OCA was generally well-tolerated by the trial patients, with side effects in the treatment groups not meaningfully different than those reported on placebo (apart from mild constipation in the 50 mg group). Consistent with anticipated FXR-related lipid metabolic effects starting with the clearance of excess lipid load from the liver, there were changes in mean serum lipid profiles observed in the OCA treatment groups compared with the placebo group that included decreased concentrations of triglycerides, increased concentrations of LDL-C and slightly decreased concentrations of HDL-C from baseline. In its publication of the results, the company observed that once-daily treatment for six weeks at the 25 mg OCA dose, which the company subsequently selected to advance in its NASH development program, led to an approximately 12% decrease in mean triglycerides to 170 mg/dL from a baseline mean level of 193 mg/dL, an approximately 22% increase in mean LDL cholesterol to 120 mg/dL from a baseline mean level of 98 mg/dL, and an approximately 5% decrease in mean HDL cholesterol to 35 mg/dL from a baseline mean level of 37 mg/dL.
(b) Phase 2 FLINT Trial for NASH
OCA achieved the primary endpoint in a Phase 2b clinical trial for the treatment of NASH, known as the FLINT trial, which was sponsored by the U.S. National Institute of Diabetes and Digestive and Kidney Diseases, or NIDDK, a part of the National Institutes of Health. A significantly greater number of OCA-treated patients also achieved an improvement of at least one fibrosis stage (35% vs 19%, p = 0.004), with OCA showing greater response rates as compared to placebo across all stages of fibrosis. The results from the FLINT trial were published in the Lancet in November 2014. This trial was a double-blind, placebo-controlled trial of a once-daily dose of 25 mg of OCA or placebo given for 72 weeks in 283 patients with biopsy-proven NASH.
![]() Primary Endpoint
Primary Endpoint
The percentage of patients meeting the FLINT primary histological endpoint, based on liver biopsies, was defined as a decrease in the NAFLD Activity Score, or NAS, of at least two points with no increase in the fibrosis score following 72 weeks of treatment, was 45% in the OCA treatment group and 21% in the placebo group (p = 0.0002, n = 219). The mean pre-treatment baseline NAS for patients in the OCA treatment group was 5.3 of a total possible score of eight (comprised of hepatocellular ballooning 0 – 2, lobular inflammation 0 – 3 and steatosis 0 – 3). Subgroup analyses showed significant response rates in the OCA treatment group in patients with risk factors for disease progression, including baseline fibrosis stage, co-morbid type 2 diabetes mellitus, alanine transaminase, or ALT, insulin resistance and severe obesity (each factor p < 0.05 for OCA compared to placebo based on 95% confidence interval of published odds ratios). The graph below shows the results of the primary endpoint in the FLINT trial and the improvements in NAS for various subgroups published in the Lancet.
Primary Endpoint: Improvement in NAS by ≥ Two Points with no Worsening of Fibrosis
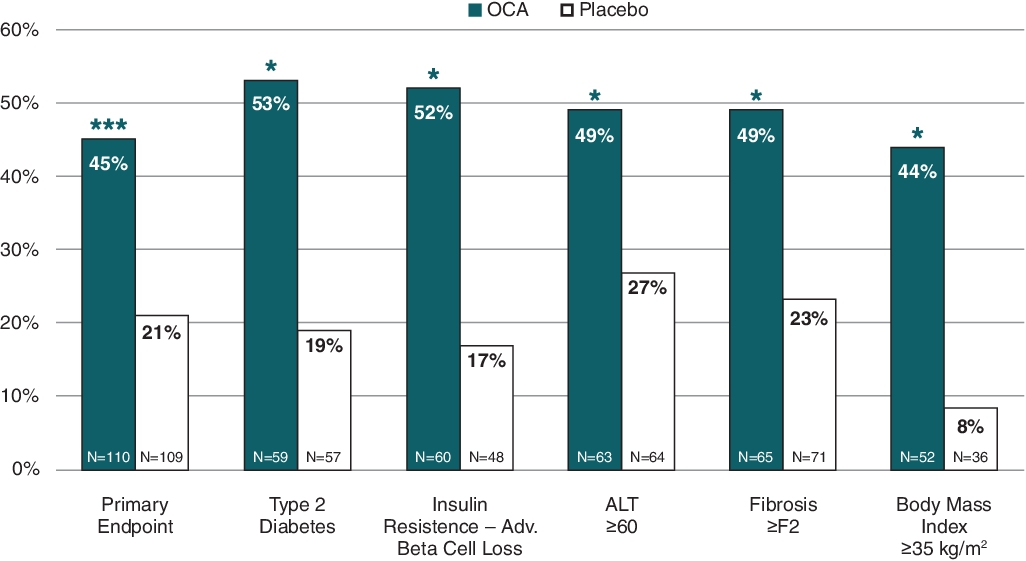
(ii) Secondary Efficacy Endpoint: Fibrosis Improvement
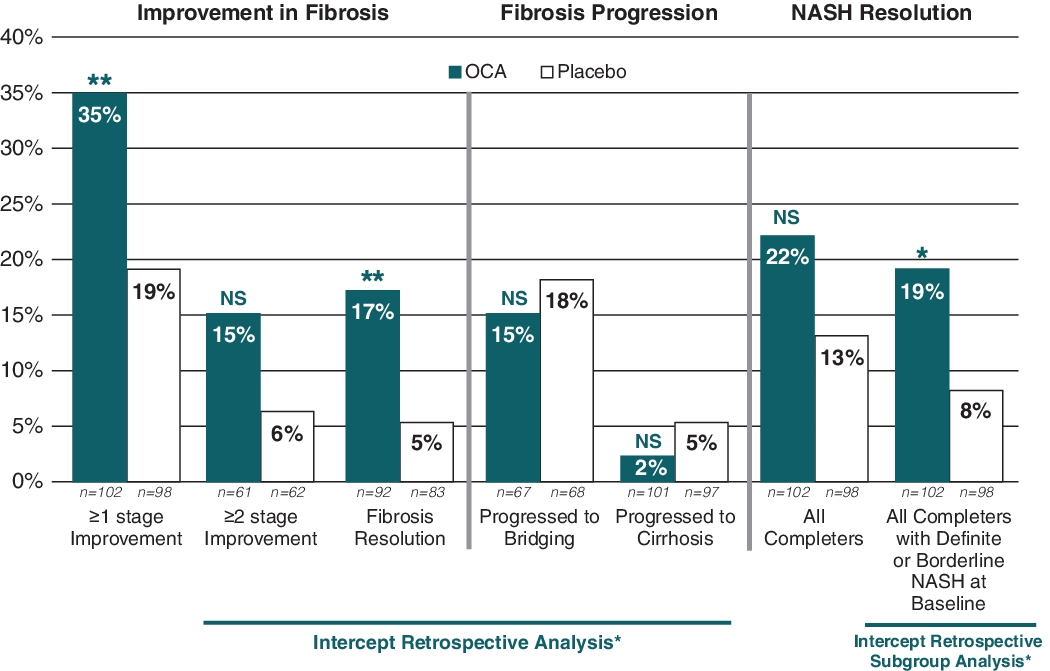
A significantly greater number of OCA-treated patients also achieved an improvement of at least one fibrosis stage (35% versus 19%, p = 0.004). Based on its retrospective analyses of the FLINT data, more OCA-treated patients exhibited fibrosis improvement of at least two fibrosis stages (15% versus 6%, not significant) and exhibited fibrosis improvements regardless of baseline fibrosis stage and a significantly greater number of OCA-treated patients also achieved complete resolution of fibrosis (17% versus 5%, p = 0.0018). Also, its retrospective analysis of the FLINT data showed that fewer OCA-treated patients progressed to bridging fibrosis (15% versus 18%, not significant) or to cirrhosis (2% versus 5%, not significant). The NASH clinical research network fibrosis staging system was used to categorize the pattern of fibrosis and architectural remodeling of the liver: no fibrosis (F0), perisinusoidal or periportal fibrosis (F1), perisinusoidal and periportal fibrosis (F2), bridging fibrosis (F3) and cirrhosis (F4). Fibrosis sub-stages 1a, 1b and 1c were considered F1 for the analysis.
(iii) Secondary Efficacy Endpoint: NASH Resolution
The secondary endpoint of NASH resolution, based on a global histological assessment, also showed improvement, although not statistically significant (22% versus 13%, p = 0.0832, not significant). A central reading of all baseline and end-of-trial biopsies was performed at the end of the trial, based on which only 80% of patients were confirmed to have definite NASH, while the remaining 20% were diagnosed as borderline NASH (10%) or not-NASH (10%). A retrospective subgroup analysis on the completer population comprised only of definite NASH patients at baseline showed that a significantly greater number of OCA-treated patients achieved NASH resolution compared with placebo-treated patients (19% versus 8%; p = 0.0278).
The graph below shows these results from the FLINT trial for fibrosis improvement, fibrosis resolution, fibrosis progression and NASH resolution.
FLINT Trial: Improvement in Histological Endpoints
(iv) Additional Secondary Endpoints
More OCA-treated patients experienced significant improvements in the major histological features of NASH, including steatosis (61% versus 38%, p = 0.001), lobular inflammation (53% versus 35%, p = 0.006) and hepatocellular ballooning (46% versus 31%, p = 0.03), as compared to the placebo treatment group. Trends were similar between the two treatment groups for portal inflammation, which is not a component of NAS and is typically mild in adult NASH patients.
The histological improvements observed in OCA-treated patients versus placebo were accompanied by significant reductions in relevant biochemical parameters, including the serum liver enzymes ALT (p < 0.0001), AST (p = 0.0001) and GGT (p < 0.0001), each of which were above generally accepted normal limits at baseline, and total bilirubin (p = 0.002). A modest but statistically significant increase in alkaline phosphatase, or ALP (p < 0.0001) in the OCA treatment group was also observed, but levels remained within typical normal limits.
OCA treatment was associated with serum lipid changes, including average increases in total cholesterol and LDL-C and an average decrease in HDL-C, that developed within 12 weeks of treatment initiation, then began reversing through the end of treatment and returned to baseline during the 24-week post-treatment follow-up phase. Based on these observations, lipid management was emphasized partway into the trial, using generally accepted guidelines. At 72 weeks as compared to baseline, the following effects were observed in the OCA treatment group: an increase in mean total cholesterol (0.16 mmol/L or 6 mg/dL increase OCA versus 0.19 mmol/L or 7mg/dL decrease placebo, p < 0.0009), an increase in mean LDL-C (0.22 mmol/L or 9 mg/dL increase OCA versus 0.22 mmol/L or 8 mg/dL decrease placebo, p < 0.0001), a decrease in mean HDL-C (0.02 mmol/L or 1 mg/dL decrease OCA versus 0.03 mmol/L or 1 mg/dL increase placebo, p = 0.01) and a decrease in triglycerides (0.22 mmol/L or 20 mg/dL decrease OCA versus 0.08 mmol/L or 7 mg/dL decrease placebo, p = 0.88, not significant).
A post-hoc analysis showed OCA-treated patients who initiated statins during the FLINT trial (n = 26) experienced a rapid reversal of their observed mean LDL-C increase to below baseline levels, with a mean decrease after 72 weeks of treatment of -18.9 mg/dL. In contrast, other OCA-treated patients with no reported initiation or change in statin therapy experienced an increase in LDL-C that peaked at week 12 and was sustained over the 72 week treatment period. Patients treated with statins at baseline who maintained statin treatment over the duration of the study (n = 50) experienced a mean LDL-C increase of 8.7 mg/dL at 72 weeks. Patients not treated with statins during the study (n = 65) experienced a mean LDL-C increase of 16.0 mg/dL. Treatment related LDL-C increases in all groups reversed with treatment discontinuation. This analysis suggests that the OCA-associated LDL-C increase reaches a maximum peak and plateaus soon after initiation of therapy and that concomitant statin use in NASH patients receiving OCA may mitigate treatment-related LDL-C increases.
In the FLINT trial, statistically significant weight loss of an average of 2.3 kilograms was observed in OCA patients compared to no weight loss in the placebo group (p = 0.008), and this weight loss reverted towards baseline during the 24-week follow-up phase. A pre-specified sensitivity analysis conducted by the investigators showed that weight loss was not a driver of the primary endpoint. An increase in a marker of hepatic insulin resistance known as HOMA-IR (calculated using the product of fasting plasma insulin and glucose) was observed at 72 weeks in the OCA treatment group (p = 0.01). However, there was an imbalance in baseline plasma insulin levels (201 pmol/L OCA versus 138 pmol/L placebo), and an even larger relative and absolute increase in HOMA-IR was observed in the placebo group at the conclusion of the 24-week follow-up phase. This is potentially attributable to the inherent variability in HOMA-IR measurements, particularly in patients with type 2 diabetes, that have been shown to make single time-point to time-point changes of this magnitude clinically uninterpretable. There were virtually no changes in mean hemoglobin A1c, a measure of average blood sugar control over a period of approximately three months, in either OCA or placebo groups at 72 weeks. In a previous study of OCA in diabetic NAFLD patients, described in more detail above, employing the hyperinsulinemic-euglycemic insulin clamp, the gold standard for detecting changes in insulin resistance, OCA improved the glucose disposal rate consistent with reduced insulin resistance.
(v) Safety and Tolerability
OCA was generally well tolerated in the FLINT trial. Adverse events were generally mild to moderate in severity and the incidence in the OCA and placebo treatment groups was similar for all symptoms except pruritus. Pruritus in the OCA treatment group occurred more frequently (23% versus 6%, p < 0.0001), at a higher grade (predominantly moderate pruritus) but resulted in only one patient discontinuation. The incidence of severe or life threatening events was not different between the two treatment groups and most of the events in both groups were deemed to be unrelated to treatment, including all severe or life threatening cardiovascular events. As previously disclosed, two deaths occurred in the OCA treatment group, but neither was considered related to OCA treatment.
(c) Phase 2 Sumitomo Dainippon Trial for NASH
In October 2015, the company announced the results of a 72-week Phase 2 dose ranging trial of OCA in 200 adult patients with NASH in Japan. The trial was conducted by its collaborator, Sumitomo Dainippon. In this trial, 202 Japanese biopsy-proven NASH patients (NAS of 5-8) were randomized into one of four arms to receive either a 10mg, 20mg or 40mg dose of OCA, or placebo, and 200 of these patients — 50 per group — initiated treatment for a 72-week double-blind treatment phase, followed by a 24-week off treatment phase. The primary endpoint was histologic improvement defined as at least a two point improvement in NAS with no worsening of fibrosis.
The primary efficacy analysis was conducted on an intention to treat, or ITT, basis, testing the dose dependent effects of once daily OCA (10 mg, 20 mg and 40 mg) versus placebo on the primary endpoint. The ITT analysis included all randomized patients who received treatment (50 per group), and patients who discontinued or did not have a repeat biopsy were treated as non-responders. A pre-specified completer analysis was conducted on the patients who had biopsies at both baseline and 72 weeks (45, 44, 44 and 37 patients in the placebo, 10 mg, 20 mg and 40 mg OCA groups, respectively).
This trial did not meet statistical significance for the primary endpoint. The ITT results in the table below show a dose dependent increase in the percentage of OCA-treated patients compared to placebo who achieved the primary endpoint (p = 0.053, not significant). The 40 mg OCA dose group achieved statistical significance on the primary endpoint compared to placebo (p = 0.0496). Dose-dependent trends not reaching statistical significance were also observed for several other pre-specified histologic endpoints, including the percentage of patients with steatosis and inflammation improvement, ballooning resolution and NASH resolution. No difference was seen in fibrosis improvement in the OCA groups compared to placebo.
| ITT Results | Placebo | 10 mg | 20 mg | 40 mg | |
|---|---|---|---|---|---|
| N = 50 | N = 50 | N = 50 | N = 50 | ||
| NAS improvement = 2 points with no worsening of fibrosis | 10 (20)% | 11 (22)% | 14 (28)% | 19 (38)% | p = 0.053 |
| p = 0.8070 | p = 0.3378 | p = 0.0496 |
In the completer analysis, similar dose dependent effects were observed, with 51% of patients in the 40 mg dose group compared to 22% in the placebo group meeting the primary endpoint (p = 0.0061).
With the exception of dose dependent pruritus, OCA appeared to be generally safe and well tolerated. The number of pruritus associated discontinuations were 0, 0, 2 and 5 patients in the placebo, 10 mg, 20 mg and 40 mg OCA groups, respectively. Changes in lipid parameters, including LDL-C, HDL-C and triglycerides, appeared to be consistent with previously reported lipid changes in Western NASH patients. No other meaningful differences in the rate of adverse events between the OCA and placebo groups were noted.
(d) Phase 2 CONTROL Trial
In December 2015, the company initiated the Phase 2 clinical trial, known as the CONTROL trial, to characterize the lipid metabolic effects of OCA and cholesterol management effects of concomitant statin administration in NASH patients.
CONTROL enrolled 80 NASH patients who were naïve to statin therapy or had undergone a statin washout period. The study included a 16-week double-blind phase followed by an optional two-year long-term safety extension phase.
At the end of the 16-week double-blind treatment period, the CONTROL trial met its primary endpoint in July 2017 by showing that newly initiated treatment with atorvastatin rapidly reversed OCA-associated increases in LDL to below baseline levels. Most of the effect was observed four weeks after initiation of the lowest available dose of atorvastatin and was sustained throughout the study period.
During the long-term safety extension phase of CONTROL, there has been one patient death. The principal investigator determined that the events leading to the patient’s death were unlikely related to OCA.
(e) Phase 3 REGENERATE Trial
Intercept Pharmaceuticals is currently conducting a Phase 3 clinical trial in non-cirrhotic NASH patients with liver fibrosis, known as the REGENERATE trial.
The REGENERATE trial was designed following discussions with the FDA and EMA. The study population is expected to primarily be comprised of Western NASH patients with histologic evidence of stage 2 or stage 3 liver fibrosis. In addition, the trial will include an exploratory cohort of NASH patients with histologic evidence of early stage 1 liver fibrosis and concomitant diabetes, obesity or elevated ALT, who are at increased risk of disease progression to cirrhosis. These patients with early stage 1 liver fibrosis will not be included in the primary endpoint analysis.
REGENERATE is designed as a double-blind, placebo-controlled Phase 3 clinical trial and is expected to enroll approximately 2,000 NASH patients at more than 350 qualified study sites worldwide and assess the potential benefits of OCA treatment on liver-related and other clinical outcomes. Patients are being randomized into one of three groups receiving a once-daily dose of placebo, 10 mg OCA or 25 mg OCA.
REGENERATE includes a pre-planned interim histology analysis after 72 weeks of treatment in patients with stage 2 or 3 liver fibrosis. If successful, the interim analysis for REGENERATE is intended to serve as the basis for seeking initial U.S. and international marketing approvals of OCA for the treatment of NASH patients with liver fibrosis. The REGENERATE trial will remain blinded after the interim analysis and continue to follow patients until the occurrence of a pre-specified number of adverse liver-related clinical events, including progression to cirrhosis, to confirm clinical benefit on a post-marketing basis.
In February 2017, the company announced modifications to the REGENERATE trial primary endpoint. Based on discussions with the FDA, the primary endpoint for the interim analysis for REGENERATE may be achieved based on one of: ![]() the proportion of OCA-treated patients relative to placebo achieving at least one stage of liver fibrosis improvement with no worsening of NASH (defined as no increase in hepatocellular ballooning or lobular inflammation) or (ii) the proportion of OCA-treated patients relative to placebo achieving NASH resolution with no worsening of liver fibrosis. Prior to this modification of the interim analysis, each of the two endpoints was required to be achieved as a co-primary endpoint. Furthermore, the company selected a definition for NASH resolution for the trial, which defines a responder as a patient achieving a histologic score of 0 for ballooning and 0 or 1 for inflammation.
the proportion of OCA-treated patients relative to placebo achieving at least one stage of liver fibrosis improvement with no worsening of NASH (defined as no increase in hepatocellular ballooning or lobular inflammation) or (ii) the proportion of OCA-treated patients relative to placebo achieving NASH resolution with no worsening of liver fibrosis. Prior to this modification of the interim analysis, each of the two endpoints was required to be achieved as a co-primary endpoint. Furthermore, the company selected a definition for NASH resolution for the trial, which defines a responder as a patient achieving a histologic score of 0 for ballooning and 0 or 1 for inflammation.
As a result of these changes, the company anticipate that the interim analysis cohort for REGENERATE will consist of approximately 750 NASH patients with stage 2 or 3 fibrosis. In May 2017, the company completed enrollment of the interim analysis cohort for the REGENERATE trial and the company anticipate top-line results from the interim analysis in the first half of 2019. The company currently intend to seek initial U.S. and international marketing approvals of OCA for the treatment of NASH patients with liver fibrosis based on the interim results from its REGENERATE trial.
In a retrospective analysis of data from the FLINT trial conducted in a REGENERATE-matched patient cohort, approximately 43% of OCA-treated patients as compared to approximately 21% of patients on placebo, achieved at least a one stage improvement in liver fibrosis without any worsening of NASH (p=0.0059). In a similar retrospective analysis on the FLINT data using the definition the company selected for NASH resolution, approximately 20% of OCA-treated patients, as compared to approximately 6% of patients on placebo achieved NASH resolution with no worsening of fibrosis (p = 0.0289).
(f) Phase 3 REVERSE Trial
Intercept Pharmaceuticals has initiated a Phase 3 trial in NASH patients with cirrhosis, known as the REVERSE trial. REVERSE is a randomized, double-blind, placebo-controlled, multi-center study to evaluate the efficacy and safety of OCA in approximately 540 patients with a biopsy-confirmed diagnosis of cirrhosis due to NASH.
The primary endpoint of the study is the percentage of subjects with histological improvement in fibrosis by at least one stage using the NASH Clinical Research Network scoring system after 12 months of treatment. Patients are being randomized in a 1:1:1 ratio to one of the three treatment arms: once-daily dosing of OCA 10 mg, once-daily OCA 10 mg with titration to 25 mg at three months, or placebo. Patients who successfully complete the double-blind phase of REVERSE will be eligible to enroll in an open-label extension phase for up to 12 additional months.
Primary Sclerosing Cholangitis (PSC)
PSC is a rare, serious, chronic cholestatic liver disease characterized by a progressive, autoimmune-based destruction of bile ducts with eventual onset of cirrhosis and its complications.
PSC is usually diagnosed by preliminary assessment of liver biochemistry, with or without reported symptoms, and confirmed by cholangiography, typically magnetic resonance cholangiopancreatography or endoscopic retrograde cholangiopancreatography, or ERCP. ALP is elevated in most PSC patients, consistent with cholestasis, and ALT and GGT are also typically elevated, but not in all cases. Bilirubin is often normal in early-stage PSC but increases with progression of the disease. The mean age at diagnosis is approximately 40 years. Approximately 75% of PSC patients have overlapping inflammatory bowel disease, principally ulcerative colitis.
Median survival for PSC patients has been previously estimated as 8 to 12 years from diagnosis in symptomatic patients, depending upon stage of the disease at the time of diagnosis. Complications involving the biliary tree are common and include cholangitis as well as ductal strictures and gallstones, both of which may require frequent endoscopic or surgical interventions. PSC is often complicated by the development of malignancies, with cholangiocarcinoma being the most common.
Despite evaluation of multiple investigational treatments, liver transplant is currently the only treatment shown to improve clinical outcomes. Ursodiol is often used for the treatment of PSC due to improvements in liver biochemistry following initiation of therapy. Despite general biochemical improvement, ursodiol has not been shown to improve transplant-free survival and, at high doses, has been associated with increased risk for serious complications. However, as there are no approved drugs for the treatment of PSC, some physicians treat patients with ursodiol, typically at a dose of 13 to 15 mg/kg/day. PSC is the fourth leading indication for liver transplant. However, the post-transplant recurrence rate of PSC has been shown to be as high as 20%.
Phase 2 AESOP Trial: OCA as Therapy in PSC
In July 2017, the company announced top-line results from an international Phase 2 clinical trial, known as the AESOP trial, to evaluate the effects of 24 weeks of treatment with varying doses of OCA compared to placebo in patients with PSC. The primary endpoint was the reduction of serum ALP levels, as compared to placebo. In addition, OCA’s effect on other secondary liver function endpoints, as well as symptoms of ulcerative colitis (a disease occurring in the majority of patients with PSC) was assessed. In October 2017, additional results from the AESOP trial were presented. OCA achieved the primary endpoint of the AESOP trial: patients receiving 5 mg of OCA daily with the option to titrate to 10 mg achieved a statistically significant reduction in ALP as compared to placebo at week 24 (p < 0.05). Patients were randomized to one of three treatment groups: placebo, OCA 1.5 – 3 mg and OCA 5 – 10 mg (with dose titration occurring at the 12-week midpoint).
| (U/L) | Placebo | OCA 1.5 – 3 mg | OCA 5 – 10 mg |
|---|---|---|---|
| (N = 25) | (N = 25) | (N = 26) | |
| Mean Baseline ALP | 563 | 423 | 429 |
| Least Squares (LS) Mean Change from Baseline in ALP at Week 12 | -53 | -57 | -135 |
| LS Mean Change from Baseline in ALP at Week 24 | -27 | -105 | -110 |
| LS Mean Percent Change from Baseline at Week 24 | 1 | -22 | -22 |
Patients in the OCA 1.5 – 3 mg group achieved statistically significant reductions in ALP versus placebo as measured by least square, or LS, mean percent change from baseline at week 24. By week 24, ALP increased 1% in the placebo group and decreased by 22% in both the OCA 1.5 – 3 mg and OCA 5 – 10 mg groups (p < 0.05).
In AESOP, a significant proportion of patients used ursodiol, with 48%, 48% and 46% of patients on placebo, OCA 1.5 – 3 mg and OCA 5 – 10 mg, respectively, receiving ursodiol at baseline. In a post-hoc analysis examining the effects of OCA in the presence and absence of ursodiol, ALP reductions were observed with OCA regardless of treatment with ursodiol. Patients receiving OCA monotherapy had greater reductions in ALP at week 12 and at week 24 as compared to patients who received OCA in addition to ursodiol. At week 12, patients in the OCA 5 – 10 mg group receiving OCA monotherapy achieved a 30% LS mean reduction in ALP as compared to a 16% reduction in patients receiving OCA in combination with ursodiol. At week 24, LS mean reductions in ALP in the OCA 5 – 10 mg group were 25% for patients receiving OCA monotherapy and 14% for patients receiving OCA in combination with ursodiol.
Pruritus is a common symptom of PSC and was the most common adverse event observed in AESOP, occurring in 46%, 60% and 67% of patients in the placebo, OCA 1.5 – 3 mg and OCA 5 – 10 mg groups, respectively.
Following the completion of the 24-week double-blind portion of the trial, patients were given the option to enroll in an open-label, long-term safety and efficacy extension trial. Of those patients who completed the double-blind phase of the AESOP trial, 97% chose to participate in the open-label extension phase.
Biliary Atresia
Biliary atresia is a life-threatening condition in infants in which the bile ducts inside or outside the liver do not have normal openings. With biliary atresia, bile becomes trapped, builds up, and damages the liver. The damage leads to scarring, loss of liver tissue, and cirrhosis. The two types of biliary atresia are fetal and perinatal. Fetal biliary atresia appears while the baby is in the womb. Perinatal biliary atresia is much more common and does not become evident until two to four weeks after birth. Some infants, particularly those with the fetal form, also have birth defects in the heart, spleen, or intestines. Biliary atresia is rare and only affects about one out of every 18,000 infants. The disease is more common in females, premature babies, and children of Asian or African American heritage. Biliary atresia is not an inherited disease and is most likely caused by an event in the womb or around the time of birth. No single test can definitively diagnose biliary atresia, resulting in the need for a series of tests. All infants who still have jaundice two to three weeks after birth, or who have gray or white stools after two weeks of birth, should be checked for liver damage.
Once diagnosed, biliary atresia is treated with a liver transplant or, more frequently, a surgery called the Kasai procedure, in which the bile ducts are connected directly to the small intestine. After the Kasai procedure, some infants continue to have liver problems and, even with the return of bile flow, some infants develop cirrhosis. Possible complications after the Kasai procedure include ascites, bacterial cholangitis, portal hypertension, and pruritus. Even after a successful Kasai surgery, most infants with biliary atresia slowly develop cirrhosis over the years and require a liver transplant by adulthood.
Phase 2 CARE Trial: OCA as Therapy in Biliary Atresia
In October 2015, the company initiated a Phase 2 clinical trial of OCA, known as the CARE trial, in pediatric patients with biliary atresia. The CARE trial will evaluate the effects of 11 weeks of OCA treatment where patients with biliary atresia are randomized to varying doses of OCA. The primary endpoint is to evaluate the pharmacokinetics and the safety and tolerability of OCA treatment. In addition, OCA’s effect on hepatobiliary indices and biomarkers will be assessed. This trial is anticipated to enroll approximately 60 patients in the United States and Europe. In addition to studying the effects of OCA treatment in biliary atresia, this trial is a part of the approved Pediatric Investigation Plan, or PIP, in support of the Marketing Authorization Application, or MAA, for OCA in PBC in the European Union.
Potential Future Product Candidates
In addition to OCA, Intercept Pharmaceuticals is developing other novel bile acid analogs targeting FXR and a second dedicated bile acid receptor called TGR5, which is a target of particular interest for the treatment of type 2 diabetes and other gastrointestinal indications. In order to streamline operating expenses, the company plan to deprioritize its development programs in these products for the foreseeable future.
INT-767
INT-767 is an orally administered dual FXR and TGR5 agonist that, like OCA, is derived from the primary human bile acid chenodeoxycholic acid, or CDCA. This product candidate has been shown to be approximately three times more potent than OCA as an FXR agonist. In animal models of chronic liver, intestinal and kidney diseases, INT-767 has consistently demonstrated greater anti-fibrotic and anti-inflammatory effects than OCA.
Intercept Pharmaceuticals has received assignments of rights to the INT-767 patent portfolio from all inventors, with the exception of one inventor. That inventor is contractually obligated to provide an assignment to it. Thus, the company believe that Intercept Pharmaceuticals is the owner of the INT-767 patent portfolio by virtue of this contractual obligation and the patent assignments Intercept Pharmaceuticals has received.
Intercept Pharmaceuticals has completed a Phase 1 clinical trial of INT-767 in healthy volunteers. The goal of the Phase 1 trial was to assess safety and pharmacokinetics in a single ascending dose escalation phase followed by a multiple ascending dose phase in healthy volunteers.
INT-777
INT-777 is an orally administered TGR5 agonist that is derived from the primary human bile acid cholic acid. Intercept Pharmaceuticals has completed preclinical studies necessary for the filing of an IND. By virtue of the patent assignments Intercept Pharmaceuticals has received and other contractual obligations owed to it, the company believe Intercept Pharmaceuticals is the exclusive owner of the INT-777 patent portfolio.
The company's in vitro studies of INT-777 showed that the product candidate has the potential to selectively target TGR5, a receptor that has been shown to directly regulate the release of glucagon like peptide-1, or GLP-1, a gastrointestinal peptide hormone with potent anti-diabetic effects. TGR5 has also been shown in animal models to regulate other metabolic pathways in brown fat and skeletal muscle that drive energy expenditure. The receptor may also play a role in the control of inflammation, which is increased in insulin resistant diabetic conditions.
In animal models of diabetes, treatment with INT-777 induced GLP-1 secretion, with resulting insulin sensitivity and normalization of glycemic control, increased basal energy expenditure and prevention of weight gain, and a reduction in blood lipid levels together with liver steatosis and fibrosis. The company believe that these preclinical results could support further development of INT-777 and its other TGR5 agonists in the treatment of type 2 diabetes, associated metabolic disorders and other gastrointestinal indications.
Strategic Collaborations and Research Arrangements
Sumitomo Dainippon Pharma
On March 29, 2011, the company entered into a license agreement with Sumitomo Dainippon, under which the company granted Sumitomo Dainippon an exclusive license to research, develop and commercialize OCA as a therapeutic for the treatment of PBC and NASH in Japan and China (excluding Taiwan). Under the terms of the agreement, Sumitomo Dainippon is required to use commercially reasonable efforts to develop and commercialize OCA in its licensed territories for the treatment of PBC and NASH, and Intercept Pharmaceuticals is obligated under the agreement to use commercially reasonable efforts to develop OCA outside of Sumitomo Dainippon’s licensed territories. Intercept Pharmaceuticals is also responsible for supplying Sumitomo Dainippon with a clinical and commercial supply of OCA requested by Sumitomo Dainippon pursuant to clinical and commercial supply agreements that include terms specified in the agreement. Sumitomo Dainippon has agreed, during the term of the agreement, to not commercialize any compound that is an FXR agonist for use in the treatment of PBC or NASH other than pursuant to the agreement.
The company granted Sumitomo Dainippon an option under the agreement to obtain an exclusive license to commercialize OCA for indications other than PBC and NASH on the same terms as are set forth in the agreement. Sumitomo Dainippon may exercise this option with respect to any indication at any time during the two-year period commencing on the date the company notify Sumitomo Dainippon of the commencement of a Phase 3 clinical trial involving OCA for such indication, subject to Sumitomo Dainippon’s payment of an option fee for each additional indication. No option fee is required to be paid by Sumitomo Dainippon if it exercises its option for any additional indication in China.
In addition to Japan and China, which are the original licensed territories, the company also granted Sumitomo Dainippon an option under the agreement to add Korea, Taiwan, Malaysia, Vietnam, the Philippines, Thailand, Singapore and/or Indonesia to its exclusive license on the same terms as are set forth in the agreement (the “Country Option”). In May 2014, Sumitomo Dainippon exercised its option to add Korea to its licensed territories.
In February 2018, the company amended its license agreement with Sumitomo Dainippon. Under the amendment, Sumitomo Dainippon agreed to return the rights to develop and commercialize OCA in Japan and Korea and the company agreed to forego any further milestone or royalty payments for the development and commercialization of OCA in such countries. In addition, Sumitomo Dainippon waived its rights to the Country Option and the parties adjusted certain milestone payment obligations with respect to the development and commercialization of OCA.
Sumitomo Dainippon may be required to pay it up to an aggregate of approximately $4 million for the achievement of development milestones, $19 million for the achievement of regulatory approval milestones and tiered royalties up to the mid-twenties in percentage terms based on net sales of OCA products in China. The term of the agreement, and Sumitomo Dainippon’s obligation to pay royalties to it for each OCA product, expires in China on the later of the expiration of the exclusivity period in China, whether through the expiration of applicable patents or the introduction of generic drugs that compete with the OCA product, or ten years after the first commercial sale of such OCA product for the first or second indication in China. Under the amendment, the parties also agreed that if certain clinical development milestones in China are not met by December 31, 2020, Sumitomo Dainippon may choose either to pay the Company a milestone payment or terminate the Agreement.
Royalty rates are subject to reduction under the agreement in specified circumstances, including if sales of generic products reach a certain threshold market share in China over a specified period.
Sumitomo Dainippon may terminate the agreement in its entirety, with respect to China or on an indication-by-indication basis upon 90 days’ written notice. Either the company or Sumitomo Dainippon may terminate the agreement in the event of the uncured material breach by or bankruptcy of the other party, subject to certain dispute resolution procedures. If Sumitomo Dainippon were to terminate the agreement for its material breach, it would have a perpetual license following the effective date of termination, subject to the payment by Sumitomo Dainippon of a royalty based on net sales of OCA products, the amount of which will depend on whether the effective date of termination occurs prior to or after the date of first commercial sale of an OCA product. If the company were to terminate the agreement for Sumitomo Dainippon’s material breach or if Sumitomo Dainippon were to voluntarily terminate the agreement, Sumitomo Dainippon’s license under the agreement would terminate.
Competition
The biopharmaceutical industry is characterized by intense competition and rapid innovation. The company's potential competitors include major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies and universities and other research institutions. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. The company believe the key competitive factors that will affect the development and commercial success of its product candidates are efficacy, safety and tolerability profile, reliability, convenience of dosing, price, the level of generic competition and reimbursement.
The company compete, and will continue to compete, with existing and new products being developed by its competitors. Some of these competitors are pursuing the development of pharmaceuticals that target the same disease and conditions that its research and development programs target.
For example, Ocaliva competes with ursodiol, a first line therapy that is approved for treatment and is generically available at a significantly lower cost than branded products.
Ocaliva is an FXR agonist. Intercept Pharmaceuticals is aware of several other companies that have FXR agonists in Phase 2 or earlier clinical or preclinical development for the treatment of PBC, including, FXR agonists from Novartis International AG (LJN452), Gilead Sciences, Inc. (GS-9674) and Enanta Pharmaceuticals, Inc. (EDP-305). Additional product candidates in Phase 2 or earlier clinical or preclinical development for the treatment of PBC include Genfit SA’s dual PPAR alpha/delta agonist (elafibranor), Cymabay Therapeutics, Inc.’s PPAR delta agonist (seladelpar), Arena Pharmaceuticals (ADP334), Bristol-Myers Squibb’s marketed anti-CTL4 fusion protein (abatacept), and FF Pharmaceuticals’ anti-CD40 monoclonal antibody (FFP104). Additionally, several companies have product candidates aimed at the cholestatic-induced pruritus associated with PBC, including apical sodium dependent bile acid transport inhibitors being developed by GlaxoSmithKline (GSK2330672).
The use of Ocaliva to treat PBC also competes off-label with fibrates. While many fibrates are specifically contraindicated for use in PBC due to potential concerns over acute and long-term safety in this patient population. An investigator-sponsored Phase 3 trial of bezafibrate, a fibrate that has not been approved for commercialization by the FDA only available outside of the United States, has been completed. Dr. Falk Pharma GmbH is running a Phase 3 trial evaluating a combination of ursodiol and budesonide, a steroid.
With respect to NASH, there are currently no therapeutic products approved for the treatment of NASH, NAFLD, portal hypertension, complications of cirrhosis or alcoholic hepatitis. If approved, Ocaliva for the treatment of NASH, would compete with several marketed therapeutics that are currently used off-label for the treatment of NASH, such as vitamin E (an antioxidant), insulin sensitizers (e.g., metformin), antihyperlipidemic agents (e.g., gemfibrozil), pentoxifylline and ursodiol. Additionally, there are ongoing and announced Phase 3 clinical trials by its competitors for the treatment of NASH, including Genfit SA’s PPAR alpha/delta agonist (elafibranor), Gilead Sciences, Inc.’s ASK-1 inhibitor (GS-4997) and Allergan’s dual CCR2 and CCR5 inhibitor (cenicriviroc), as well as several FXR agonists in Phase 2 or earlier clinical or preclinical development for the treatment of NASH, including, FXR agonists from Novartis International AG (LJN452), Gilead Sciences, Inc. (GS-9674) and Enanta Pharmaceuticals, Inc. (EDP-305).
Additional product candidates in Phase 2 or earlier clinical or preclinical development for the treatment of NASH include Bristol-Myers Squibb Co., Novo Nordisk A/S, Conatus Pharmaceuticals Inc., Cymabay Therapeutics, Inc., Islet Sciences, Inc., Galectin Therapeutics Inc., Zydus Pharmaceuticals Inc., NGM Biopharmaceuticals Inc., Galmed Medical Research Ltd., MediciNova, Inc., Ionis Pharmaceuticals, Inc., FibroGen, Inc., Viking Therapeutics, Inc., AstraZeneca plc, Durect Corporation, Immuron Ltd., Boehringer Ingelheim GmbH, MiNA Therapeutics, NuSirt Biopharma, Inc., Protalix Biotherapeutics, and Medivation, Inc.
With respect to PSC, there is no approved treatment for PSC. If approved, Ocaliva, for the treatment of PSC, would compete off-label with ursodiol. Intercept Pharmaceuticals is also aware of several companies that have product candidates in Phase 2 clinical or earlier stage preclinical development for the treatment of PSC, including Allergan Plc, Biotie Therapies Corp. (acquired by Acorda Therapeutics, Inc.), Dr. Falk Pharma GmbH, Durect Corporation, Gilead Sciences, Inc. and Shire plc.
The company believe that OCA offers key potential competitive advantages over ursodiol and other products in development. However, many of its potential competitors have substantially greater financial, technical and human resources than the company do, as well as greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of products and the commercialization of those products. Accordingly, its competitors may be more successful than it in obtaining approval from the FDA or from other regulators for drugs and achieving widespread market acceptance. The company's competitors’ drugs may be more effective, or more effectively marketed and sold, than any product candidate the company may commercialize and may render its product candidates obsolete or non-competitive before the company can recover the expenses of their development and commercialization. The company anticipate that the company will face intense and increasing competition as new drugs enter the market and other advanced technologies become available. Finally, the development of new treatment methods for the diseases Intercept Pharmaceuticals is targeting could render its product candidates non-competitive or obsolete. NASH is a complex disease and it is unlikely that any one therapeutic option will be optimal for every NASH patient. In addition, its ability to compete may be affected because, in many cases, insurers or other third-party payors seek to encourage the use of generic products.
Intellectual Property
The proprietary nature of, and protection for, its product candidates and its discovery programs, processes and know-how are important to its business. Intercept Pharmaceuticals has sought patent protection in the United States and internationally for OCA, INT-767 and INT-777, and its discovery programs, and other inventions to which Intercept Pharmaceuticals has rights, where available and when appropriate. The company's policy is to pursue, maintain and defend patent rights, whether developed internally or licensed from third parties, and to protect the technology, inventions and improvements that are commercially important to the development of its business. The company also rely on trade secrets that may be important to the development of its business.
The company's commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of its current and future product candidates and the methods used to develop and manufacture them, as well as successfully defending these patents against third-party challenges. The company's ability to stop third parties from making, using, selling, offering to sell or importing its products depends on the extent to which Intercept Pharmaceuticals has rights under valid and enforceable patents or trade secrets that cover these activities. The company cannot be sure that patents will be granted with respect to any of its pending patent applications or with respect to any patent applications filed by it in the future, nor can the company be sure that any of its existing patents or any patents that may be granted to it in the future will be commercially useful in protecting its product candidates, discovery programs and processes. For this and more comprehensive risks related to its intellectual property, please see Item 1.A. “Risk Factors — Risks Related to The company's Intellectual Property.”
OCA (lead product candidate; FXR agonist)
The patent portfolio for OCA contains patents and patent applications directed to compositions of matter, manufacturing methods, and methods of use. As of December 31, 2017, the company owned eleven U.S. patents, thirteen pending U.S. patent applications, and corresponding foreign patents and patent applications. Foreign patents have been granted in 30 European countries as well as Australia, Canada, China, India, Israel, Japan, and Macao. The company expect the composition of matter patents, if the appropriate maintenance, renewal, annuity or other governmental fees are paid, to expire in 2022 (worldwide) at the soonest and 2033 at the latest. In conjunction with the accelerated approval of Ocaliva in the United States, Intercept Pharmaceuticals has applied to extend the 2022 expiration date of the composition of matter patent in the United States by five additional years under the provisions of the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act. Intercept Pharmaceuticals has also made a similar application in Europe in conjunction with the conditional approval of Ocaliva for a supplementary protection certificate to extend the composition of matter patent in Europe by five additional years. Patent term extension may be available in certain foreign countries upon regulatory approval. The company expect the other patents in the portfolio, if the appropriate maintenance, renewal, annuity, or other governmental fees are paid, to expire from 2022 to 2033.
INT-767 (dual FXR/TGR5 agonist)
The patent portfolio for INT-767 contains patents and patent applications directed to compositions of matter and methods of use. As of December 31, 2017, the company owned five U.S. patents, seven pending U.S. patent applications, and corresponding foreign patents and patent applications. Foreign patents have been granted in Australia, Canada, China, 32 European countries as well as Hong Kong, India, Israel and Japan. The company expect the issued composition of matter patent in the United States, if the appropriate maintenance, renewal, annuity or other governmental fees are paid, to expire in 2029. It is possible that the term of the composition of matter patent in the United States may be extended up to five additional years under the provisions of the Hatch-Waxman Act. The company expect the foreign composition of matter patents, if the appropriate maintenance, renewal, annuity or other governmental fees are paid, to expire in 2027. Patent term extension may be available in certain foreign countries upon regulatory approval. The company expect the other patents in the portfolio, if the appropriate maintenance, renewal, annuity or other governmental fees are paid, to expire from 2027 to 2029.
INT-777 (TGR5 agonist)
The patent portfolio for INT-777 contains patents and patent applications directed to compositions of matter and methods of use. As of December 31, 2017, the company owned five U.S. patents, one pending U.S. patent applications, and corresponding foreign patents and patent applications. Foreign patents have been granted in Australia, China, 9 Eurasian countries, 30 European countries, Hong Kong, Israel, Japan, Macao, Mexico, Singapore, South Korea, and South Africa. The company expect the composition of matter patent in the United States, if the appropriate maintenance, renewal, annuity or other governmental fees are paid, to expire in 2030. It is possible that the term of the composition of matter patent in the United States may be extended up to five additional years under the provisions of the Hatch-Waxman Act. The company expect the foreign composition of matter patents, if the appropriate maintenance, renewal, annuity or other governmental fees are paid, to expire beginning in 2028. Patent term extension may be available in certain foreign countries upon regulatory approval. The company expect the other patents in the portfolio, if the appropriate maintenance, renewal, annuity or other governmental fees are paid, to expire from 2028 to 2030.
Trade Secrets
In addition to patents, the company rely on trade secrets and know-how to develop and maintain its competitive position. Trade secrets and know-how can be difficult to protect. The company seek to protect its proprietary processes, in part, by confidentiality agreements and invention assignment agreements with its employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect its proprietary information. The company also seek to preserve the integrity and confidentiality of its data, trade secrets and know-how by maintaining physical security of its premises and physical and electronic security of its information technology systems.
Manufacturing
The company do not own or operate manufacturing facilities for the production of Ocaliva or any of its product candidates, nor do Intercept Pharmaceuticals has plans to develop its own manufacturing operations in the foreseeable future. The company currently rely on third-party contract manufacturers for all of its required raw materials, active pharmaceutical ingredient, or API, and finished product for commercial sales and for its clinical trials and preclinical studies.
The company currently have a commercial manufacturing and supply agreement with PharmaZell GMBH, or PharmaZell that the company entered into in September 2016. Under the agreement Intercept Pharmaceuticals has agreed to purchase from PharmaZell a certain percentage of its annual commercial requirements of API for use in Ocaliva. The company also agreed to purchase specified minimum quantities of API. The agreement is effective until December 31, 2020, subject to two-year automatic renewal terms, unless either party provides notice of non-renewal at least 12 months prior to the end of the initial term or the then-current renewal term. The company may terminate the agreement immediately with written notice upon the occurrence of certain regulatory events, or if PharmaZell fails to meet certain quality standards, applicable laws or specified delivery obligations. Each party also has the right to terminate the Agreement immediately upon written notice for other customary reasons such as material breach and bankruptcy.
Intercept Pharmaceuticals is currently seeking to qualify one or more back-up API manufacturers and the company do not have any current contractual relationships for the manufacture of commercial supplies of any of its products or product candidates other than OCA. The company currently obtain these supplies and services from its third-party contract manufacturers on a purchase order basis. The company intend to enter into agreements with a third-party contract manufacturer and one or more back-up manufacturers for the commercial production of those product candidates.
Contract manufacturers are subject to extensive governmental regulation and the company depend on them to manufacture Ocaliva and its product candidates in accordance with applicable current good manufacturing practice regulations or cGMPs. Development and commercial quantities of any products that the company develop will need to be manufactured in facilities, and by processes, that comply with the requirements of the FDA, EMA and the regulatory agencies of other jurisdictions in which Intercept Pharmaceuticals is seeking approval.
Sales and Marketing
Intercept Pharmaceuticals is commercializing Ocaliva in the United States, Europe and Canada using its internal commercial organization, as well as a contract sales organization. Intercept Pharmaceuticals has built and plan to continue to expand its commercial infrastructure in Europe, Canada and Australia and will likely seek to commercialize OCA through distribution or other collaboration arrangements outside of the United States, Europe, Canada and Australia, subject to obtaining necessary marketing approvals. The company also have a team of field-based medical science liaisons, who play an important role in providing medical information about Ocaliva to clinicians and other health care professionals.




