Kitov Pharamceuticals
Company History
Kitov Pharma (KTOV) was incorporated under the laws of the State of Israel (under a previous name) on August 12, 1968 and its ordinary shares were originally listed for trading on the TASE in 1978. The company's ordinary shares are currently traded on the TASE under the symbol “KTOV”, and its ADSs and its public warrants are traded on NASDAQ under the symbols “KTOV” and “KTOVW”, respectively.1
In October 2012, the District Court in Lod, Israel approved the creditors arrangement in accordance with Section 350 of the Companies Law in order to effectuate the sale by Kitov Pharma (then known as Mainrom Line Logistics Ltd.) of all its activities, assets, rights, obligations and liabilities to a private company held by its then controlling shareholders, and all rights of Kitov Pharma’s creditors against it were extinguished. The sale was made pursuant to an arrangement between Kitov Pharma and its creditors. Following such sale and a related cash distribution to Kitov Pharma’s shareholders, Kitov Pharma remained without any assets, debt and/or liabilities. As described in the District Court approval, in connection with the sale, on October 31, 2012, the former controlling shareholders sold control of Kitov Pharma (then a shell company) to Mr. Sheer Roichman. From the completion of these transactions until the completion of the acquisition of Kitov Pharmaceuticals described below, Kitov Pharma did not conduct any business activities and was a public shell company listed on the TASE with no assets, debt and/or liabilities.
Kitov Pharma had a wholly owned Israeli subsidiary, Kitov Pharmaceuticals Ltd., which, prior to the completion of its merger with and into Kitov Pharma in December 2017, together with Kitov Pharma, was engaged in the research and development of Consensi™. Kitov Pharmaceuticals Ltd. was founded in June 2010, and pursuant to an Asset Purchase Agreement, dated October 13, 2010, between Kitov Pharmaceuticals and JPW PCH LLC, or JPW, JPW sold to Kitov Pharmaceuticals JPW’s rights and interests in and to U.S. and international patent applications relating to Consensi™ and KIT-301, which was a combination drug that the Company subsequently determined to remove from its development pipeline. Kitov Pharmaceuticals assumed all liabilities arising from ownership, use or exercise, of rights under, the patent applications.
On July 11, 2013, Kitov Pharma acquired Kitov Pharmaceuticals Ltd. As part of the acquisition, Mainrom Line Logistics Ltd. changed its name to Kitov Pharmaceuticals Holdings Ltd., which name was subsequently changed in January 2018 to Kitov Pharma Ltd.
On November 25, 2015, Kitov Pharma completed an initial public offering on NASDAQ of ADSs and public warrants to purchase ADSs. The gross proceeds to it from this offering were approximately $13 million, prior to deducting underwriting discounts, commissions and other offering expenses.
On January 13, 2017, the company announced that the company had acquired a majority equity stake in TyrNovo Ltd., a privately held developer of novel small molecules in the oncology therapeutic field. For more information, see, “Item 4. Information on the Company – History and Development of the Company – Recent Developments - Acquisition of TyrNovo” below.
On April 25, 2017, the boards of directors of each of Kitov Pharma and Kitov Pharmaceuticals approved a merger between the two entities, with Kitov Pharma remaining as the surviving entity. The merger was completed in December 2017. Kitov Pharmaceuticals was dissolved upon the merger, and Kitov Pharma remained as the surviving entity. For more information on the merger, see Item 4.C – Organizational Structure.
The company had no material capital expenditures for the years ended December 31, 2017, 2016, and 2015.
Recent Developments
Acquisition of TyrNovo
In January 2017, the company announced that Kitov Pharma acquired a majority equity stake in TyrNovo Ltd., a privately held developer of novel small molecules in the oncology therapeutic field, for consideration of approximately $2 million in cash and $1.8 million equivalent of Kitov Pharma’s ordinary shares based on the closing price of Kitov Pharma’s shares on the TASE on January 11, 2017, or 11,292,508 ordinary shares. In October 2017, the company announced the acquisition of an additional 27% stake in TyrNovo from unaffiliated minority shareholders of TyrNovo who collectively held 4,024 ordinary shares (the “Newly Acquired TyrNovo Shares”). In exchange for the Newly Acquired TyrNovo Shares, Kitov Pharma will issue to these unaffiliated minority shareholders of TyrNovo, in aggregate, 13,169,689 newly issued ordinary shares (equivalent to 658,484 ADSs) of Kitov Pharma. After the closing of this new share exchange transaction, which is pending receipt by the selling TyrNovo shareholders of a tax ruling from the Israeli Tax Authority and is expected to close by March 15, 2018, and assuming no other issuances of equity by TyrNovo until such time, the company will hold approximately 91.9% of TyrNovo’s issued and outstanding ordinary shares. Approximately 3.9% of TyrNovo’s ordinary shares are owned by Dr. Hadas Reuveni Ph.D., the founder and Chief Technology Officer of TyrNovo. An additional approximately 4.1% of TyrNovo’s ordinary shares are owned by Taoz – Company for Management and Holdings of Companies Ltd. (“Taoz”), a minority shareholder with whom Kitov Pharma entered into a shareholders’ agreement in February 2017.
TyrNovo is developing NT219, a small molecule that presents what the company believe to be a new concept in cancer therapy by affecting two key oncology-related mechanisms, Insulin Receptor Substrates (IRS) 1 and 2, as well as the Signal Transducer and Activator of Transcription 3 (STAT3). In pre-clinical trials in PDX models, NT219, in combination with several approved oncology drugs, displayed potent anti-tumor effects in various cancers by preventing the tumors from developing resistance to the approved drug treatments and re-sensitizing tumors to the approved drugs even after resistance was acquired. For more information regarding NT219, see, “Item 4. Business Overview - The company's Therapeutic Candidates – NT219”.
July 2017 Registered Direct Offering
On July 14, 2017, the company completed a registered direct offering of 2,431,746 ADSs representing 48,634,920 Ordinary Shares. In a concurrent private placement, the company sold to the purchasers of its ADSs in this registered direct offering warrants to purchase 1,215,873 ADSs, representing one-half the number of the ADSs purchased by such investors in the registered direct offering. The company will receive gross proceeds from the concurrent private placement transaction solely to the extent such warrants are exercised for cash. The warrants were initially exercisable on the six-month anniversary of the issuance date at an exercise price of $1.50 per ADS and will expire five years from the initial exercise date. The warrants and the ADSs issuable upon the exercise of the warrants were offered pursuant to an exemption from the registration requirements of the Securities Act and pursuant to Regulation S under the Securities Act. Each ADS together with a warrant to purchase one-half of an ADS was sold at a negotiated price of $1.45. The gross proceeds to it from this offering in July 2017 were approximately $3.5 million, prior to deducting placement agent fees and other offering expenses. As of the date of this Annual Report on Form 20-F, warrants to purchase 210,276 ADSs have been exercised either for cash or in cashless exercises, and to date, as a result of the cash exercises, Kitov Pharamceuticals has received additional proceeds from this offering of approximately $270,000.
Phase III/IV Renal Function Clinical Trial
Additional data from the Phase III clinical trial of Consensi™ suggested beneficial effects on renal (kidney) function, as compared to negative effects on renal function caused by other NSAIDS. For more information, see, “Item 4. Business Overview - The company's Therapeutic Candidates – Research and Development”.
The company completed a clinical trial designed to validate and better quantify these potential beneficial renal effects. The trial was designed to further explain the synergistic antihypertensive effect, where the reduction in diastolic blood pressure demonstrated with Consensi™ was greater than that observed with amlodipine besylate alone at certain times of the day. Accordingly, the company conducted a double blind, placebo controlled, clinical trial intended statistically to demonstrate Consensi’s™ effects on renal and vascular function, while providing it with data with respect to Consensi™ in addition to the data of the Phase III clinical trial, by utilizing a primary efficacy end-point in the renal function clinical trial comparable to that of the Phase III clinical trial. The trial was performed in the U.K. in three groups of 8 to 49 patients (and a total of 104 patients), with each patient treated over a total period of two weeks. Group One received a placebo, Group Two was treated with a standard drug available in the market for treating hypertension (amlodipine besylate, one of the components of Consensi™), and Group Three was treated with the two components of Consensi™ (celecoxib and amlodipine besylate). The primary efficacy endpoint of the trial was to show that Consensi™ lowers daytime systolic blood pressure by at least 50% of the reduction in blood pressure achieved in patients treated with amlodipine besylate only. Secondary endpoints included various parameters of renal function. The Phase III/IV renal function clinical trial for Consensi™ was conducted in medical centers in the United Kingdom on the basis of the approval of the British Regulatory Authority (MHRA), as well as the approvals of the relevant U.K. ethics committees. In October 2017, the company announced that the Phase III/IV renal function clinical trial, successfully met its primary efficacy endpoint. Data from the trial demonstrated that Consensi™ lowered systolic blood pressure a comparable amount to amlodipine besylate, thus meeting the trial’s primary efficacy endpoint of achieving at least 50% of the amlodipine reduction (p=0.019). The study also demonstrated that treatment with Consensi™ led to a statistically significant reduction of serum creatinine, a marker of renal function, from its baseline value (p=0.0005). In contrast, neither amlodipine besylate nor placebo lowered creatinine to a statistically significant level. When comparing the effect of Consensi™ to amlodipine besylate in lowering creatinine, it was found that Consensi™ enhanced the creatinine reduction by an average of 102% over that achieved with amlodipine besylate alone, although there was a slight, but statistically insignificant, increase in the rate of edema in the Consensi™ treatment arm. Although the Phase III/IV renal function clinical trial was not required as part of the initial Consensi™ NDA submission to the FDA, the company delivered the initial study results data to the FDA shortly following completion of the study, and the company anticipate that the company will submit the completed clinical study report to the FDA within six to eight weeks of this Annual Report on Form 20-F.
Consensi™ New Drug Application to FDA
The company submitted the New Drug Application (NDA) for marketing approval of Consensi™ to the U.S. Food and Drug Administration (FDA) in July 2017, and on October 2, 2017 the company announced that the FDA filed the NDA, thereby granting a full review. In connection with its determination that its application is sufficiently complete to permit a substantive review, the FDA, under the Prescription Drug User Fee Act (PDUFA), has set a target date of May 31, 2018 to complete its review. The company subsequently submitted the mandated four-month safety update to the NDA on November 30, 2017.
The company received a waiver from the FDA for the $2 million NDA fee for Consensi™.
Business Overview
Kitov Pharamceuticals is a development stage biopharmaceutical company currently focused on the development of:
- Consensi™, a combination drug for the simultaneous treatment of two clinical conditions: pain caused by osteoarthritis and hypertension (high blood pressure), which can be pre-existing or caused by the treatment for osteoarthritis; and
- NT219, a small molecule that uniquely targets two pathways highly involved in cancer drug resistance.
In addition, the company may consider the acquisition of therapeutic candidates at various stages of development in various therapeutic areas or currently approved drug products. The company currently have no binding agreements or commitments to complete any transaction for the possible acquisition of new therapeutic candidates or approved drug products.
The company intend to seek FDA approval for the commercialization of its therapeutic candidates, and where applicable through the Section 505(b)(2) regulatory path under the Federal Food, Drug, and Cosmetic Act of 1938, as amended. Where applicable, the company also intend to seek corresponding regulatory paths for approval in other foreign jurisdictions. The company's current pipeline consists of two therapeutic candidates: ![]() Consensi™, which successfully completed its Phase III clinical trial and which is presently subject to review and approval by the FDA, following the successful filing of a completed 505(b)(2) NDA and (ii) NT219, which is in a preclinical stage but will likely be subject to review and approval by the FDA upon filing a completed 505(b)(1) NDA, if at all. Upon and subject to receipt of the requisite approvals, the company intend to commercialize its therapeutic candidates through licensing and other commercialization arrangements with pharmaceutical companies on a global and/or territorial basis. The company may also evaluate, on a case by case basis, co-development and similar arrangements, as well as independent commercialization of its therapeutic candidates.
Consensi™, which successfully completed its Phase III clinical trial and which is presently subject to review and approval by the FDA, following the successful filing of a completed 505(b)(2) NDA and (ii) NT219, which is in a preclinical stage but will likely be subject to review and approval by the FDA upon filing a completed 505(b)(1) NDA, if at all. Upon and subject to receipt of the requisite approvals, the company intend to commercialize its therapeutic candidates through licensing and other commercialization arrangements with pharmaceutical companies on a global and/or territorial basis. The company may also evaluate, on a case by case basis, co-development and similar arrangements, as well as independent commercialization of its therapeutic candidates.
Background on Consensi™ and NT219
Consensi™ is based on the generic drugs celecoxib and amlodipine besylate. Celecoxib, the active ingredient in the branded drug Celebrex®, is a known and approved-for-use drug designed primarily to relieve pain caused by osteoarthritis. Amlodipine besylate is a known and approved-for-use drug designed to reduce blood pressure. This combination is designed to simultaneously relieve pain caused by osteoarthritis and treat hypertension, which is one of the side effects of using non-steroidal anti-inflammatory drugs, or NSAIDs, for treating pain caused by osteoarthritis.
During 2017, the company acquired a majority of the shares in TyrNovo, a privately held Israeli developer of novel small molecules in the oncology therapeutic field. TyrNovo has developed NT219, a small molecule that presents what the company believe to be a new concept in cancer therapy by affecting two key oncology-related mechanisms, Insulin Receptor Substrates (IRS) 1 and 2, as well as the Signal Transducer and Activator of Transcription 3 (STAT3). In pre-clinical trials in PDX models, NT219, administered concomitantly with several approved oncology drugs, displayed potent anti-tumor effects in various cancers by preventing the tumors from developing resistance to the approved drug treatments and re-sensitizing tumors to the approved drugs even after resistance is acquired. For more information regarding NT219, see, “Item 4. Business Overview - The company's Therapeutic Candidates – NT219”.
Competitive strengths
The pharmaceutical market is characterized by large international pharmaceutical companies that develop a wide range of products, both generic and NCEs, which operate alongside smaller companies, such as ours, that develop a specific drug or a combination of drugs. Therefore, many small companies enter into agreements with such global companies during the drug development stage in order to continue the development or marketing of the drug, taking advantage of the financial, marketing and/or other resources available to such global companies. At the same time, the global companies tend to enter into agreements with smaller companies in order to save development time and resources. The global drug sector is a highly developed market with a turnover of hundreds of billions of U.S. dollars and intense competition. Most of the drugs the company intend to develop have or are expected to have competing drugs or other therapies, developed at the same time by other companies and organizations. Kitov Pharamceuticals is therefore exposed to competition in its field of operation. Although the company believe its therapeutic candidates have advantages which its competitors’ products lack, there is a constant risk in the drug development field that a competing party will complete the development stages before Kitov Pharamceuticals is able to develop its therapeutic candidates intended for the same disease. Moreover, a constant threat in its market is presented by new drugs that have already completed all the development stages and have already entered the market and are competing with the treatments and drugs previously available on the market.
The company believe there are several advantages to the therapeutic candidates Kitov Pharamceuticals is developing, such as:
For Consensi™:
- providing a solution to the concerns of physicians who avoid prescribing an NSAID treatment for pain caused by osteoarthritis due to its cardiovascular side effects;
- reassuring physicians who are concerned that their patients who are treated for osteoarthritis will also be treated for hypertension, which is a known side effect of NSAID treatments for pain caused by osteoarthritis. This is a particular concern, as hypertension is usually not accompanied by tangible symptoms, and therefore patients may not be aware of their condition or the need to treat it;
- using one drug that also includes an active ingredient that treats hypertension either as an existing condition or as a side effect of using other drugs, ensures that the patient receives the suitable treatment for their disease and for its side effect;
- purchasing one drug as opposed to purchasing two separate drugs may lead to financial savings for patients in the U.S. by requiring payment of just one co-payment and prescription fee as opposed to a double co-payment and prescription fee. In addition, the use of one combination drug reduces the patient’s discretion with respect to whether to purchase and use only one of the drugs and provides a comprehensive dual medical treatment in one combined drug; and
- using calcium channel blockers in its therapeutic candidates as an antihypertensive. Calcium channel blockers are not included in the FDA Safety Information Release for NSAIDs co-administered with angiotensin converting enzyme inhibitors, or ACE inhibitors, or with angiotensin II receptor antagonists.
In addition to the aforementioned medical and economic advantages of Consensi™, the company believe that Consensi™ has several commercial advantages, such as reduced development time compared to the development time of new chemical entities (NCEs) and decreased risk factors in the development process. These commercial advantages derive from the fact that combination drugs are based on known materials already approved for use by the FDA. The FDA offers a shortened regulatory procedure referred to as a “505(b)(2) NDA” to approve combination drugs. This procedure may be used to file a request to approve a product that relies on the results of the safety and effectiveness trials performed for the components of the combination in the past by others and not by the submitters of the request for approval. Accordingly, the approval process in a 505(b)(2) NDA is shorter and less expensive compared to the approval process for NCEs. In addition, the use of known, proven and safe components recognized by physicians and medical organizations, and the enhanced medical effect of concurrently treating and preventing hypertension, may shorten the time and decrease the costs usually required for the acceptance of the new product in the drug marketplace.
For NT219:
NT219 is a small molecule, and small molecules typically are less expensive to develop and have less complex CMC as compared to large proteins or antibodies. In addition, NT219 has the potentially advantageous effect of:
- overcoming drug resistance acquired by cancer; and
- working in combination with multiple approved cancer therapies.
Strategy
The company's goal is to become a significant player in the development of innovative chemical drugs with a clinical and commercial added value.
Key elements of its strategy are to:
- develop its therapeutic candidates with clinical and commercial advantages and obtain approval thereof from the FDA and other foreign regulatory authorities;
- expand its line of therapeutic candidates through the acquisition or in-licensing of technologies, products and drugs intended to meet clinical needs, thereby utilizing the skills, knowledge and experience of its personnel to develop and enhance the value of additional products, and bring them to market efficiently;
- capitalize on the FDA’s 505(b)(2) regulatory pathway, or other pathways that simplify the road to an NDA submission, to obtain more timely and efficient approval of its formulations of previously approved products, when applicable;
- cooperate with third parties to both develop and commercialize therapeutic candidates in order to share costs and leverage the expertise of others; and
- enter into sub-license agreements with international companies for potential or future therapeutic candidates based on potential upfront and milestone payments, royalties and/or other marketing arrangements, depending on product and market conditions.
therapeutic candidates, “Consensi™,” and “NT219”, are described below.
Consensi™:
Background on Osteoarthritis and Hypertension
Numerous factors influence the drug market, including the aging of the general population. As life expectancy increases, the company expect that demand will increase for innovative drugs that treat diseases related to the elderly, such as osteoarthritis and hypertension.
Osteoarthritis
Arthritis means joint inflammation. The term is used to describe the pain, stiffness and/or swelling in the joints of the body where one or more bones are joined by ligaments. A normal joint provides a smooth surface enabling adjacent bones to move and glide on each other during normal motion. In contrast, an arthritic joint is one that may have varying degrees of inflammation and possibly destruction of the joint cartilage. These destructive changes preclude normal motion and cause pain.
The most common type of arthritis is called osteoarthritis and is more common with advancing age. People with osteoarthritis usually have joint pain and a decreased range of joint movement. Unlike some other forms of arthritis, osteoarthritis affects only the joints. This condition is also sometimes called degenerative joint disease. Osteoarthritis primarily affects the joint cartilage. Healthy cartilage allows bones to glide over one another and absorbs energy from the shock of physical movement. However, with osteoarthritis, the surface layer of cartilage breaks down and wears away. This allows the bony surface of the different bones under the cartilage to rub together, causing, pain, swelling, and loss of motion of the joint. Over time, affected joints may lose their normal shape. Also, bone spurs, small growths called osteophytes, may grow on the edges of the joint further impairing joint function. Thus, bits of bone or cartilage can break off and float inside the joint space, causing more pain and possible damage.
Osteoarthritis in the younger population is usually caused by traumatic injuries to the joints. In contrast, in the older population it is a more of a chronic degenerative disease process. The main symptom of osteoarthritis is pain that appears gradually, worsens with exertion, and is transiently relieved by rest.
The pain caused by osteoarthritis is described by patients as a deep pain or a burning sensation related to the joint tissues of the affected area. Osteoarthritis mainly affects the cartilage and disrupts the structural balance in the cartilage of the joint, causing the cartilage cells to increase production of new raw materials required to create cartilage, but concurrently produce enzymes that digest the cartilage.
Osteoarthritis is one of the most common diseases worldwide causing physical disabilities in adults. According to data published in the Center for Disease Control (CDC) website, an estimated 26.9 million U.S. adults in 2005 were diagnosed with osteoarthritis, of which approximately 50% suffer from hypertension. Among individuals in the U.S., it is estimated that over 40% will eventually suffer from osteoarthritis in at least one joint.
The pharmaceuticals used for treating osteoarthritis include a range of drugs. The particular choice of treatment is made according to the disease severity. These can range from acetaminophen for cases of milder severity, to diclofenac, naproxen, and celecoxib for moderate severity, up to treatment with narcotics for the most severe cases.
Various non pharmacological treatments are intended to relieve the pain caused by the disease and to preserve and improve joint function. Among these treatments are changes in the patient’s life style, namely diet, physiotherapy and exercise. The objectives of these treatments are to strengthen the muscles adjacent to the joints and increase their ranges, thereby reducing body weight, and decreasing the loads on the weight carrying joints to subsequently reduce the intensity of the pain.
In some cases, the conservative non pharmacological treatments are not sufficiently helpful. In such cases, patients typically request medical treatment. According to data published on the website of the Mayo Clinic in April 2013, the most common medical treatments are the use of analgesics, such as NSAIDs, which include enzyme inhibitors, such as COX-2. NSAIDs treat inflammation by inhibiting enzymes responsible for the initiation of the development of inflammation and subsequent pain. COX-2 enzyme inhibitors are non-steroidal drugs that treat inflammation by directly inhibiting COX-2, an enzyme responsible for the development of inflammation and subsequent pain but do not target the COX-1 enzyme. Targeting selectivity for COX-2 reduces the risk of peptic ulceration, and is the main advantage of celecoxib, rofecoxib and other members of this drug class over non COX-2 selective NSAIDs.
After several COX-2 inhibiting drugs were approved for marketing, data from clinical trials revealed that COX-2 inhibitors caused a significant increase in heart attacks and strokes, with some drugs in the class possibly having worse risks than others. See “Business - The company's Therapeutic Candidates – Competitive Treatments for Pain Caused by Osteoarthritis”.
A typical osteoarthritis treatment plan with these analgesics is as follows: ![]() initial treatment of minor osteoarthritis will begin with use of drugs such as acetaminophen; (ii) in the event that acetaminophen treatment is not effective, the physician will proceed to treatments using NSAIDs, which will begin using drugs such as ibuprofen followed by naproxen and/or other NSAIDs (more than 20 types of drugs, including COX-2 enzyme inhibitors); (iii) in cases where treatment with these drugs is ineffective, the treatment will be direct injection of steroids into the affected joint; (iv) in cases where steroid injection is ineffective, treatment by injecting hyaluronic acid (HA) into the affected joint will be considered; and (v) in the event that all the aforementioned treatments fail, the patient may consider surgical replacement of the affected joint.
initial treatment of minor osteoarthritis will begin with use of drugs such as acetaminophen; (ii) in the event that acetaminophen treatment is not effective, the physician will proceed to treatments using NSAIDs, which will begin using drugs such as ibuprofen followed by naproxen and/or other NSAIDs (more than 20 types of drugs, including COX-2 enzyme inhibitors); (iii) in cases where treatment with these drugs is ineffective, the treatment will be direct injection of steroids into the affected joint; (iv) in cases where steroid injection is ineffective, treatment by injecting hyaluronic acid (HA) into the affected joint will be considered; and (v) in the event that all the aforementioned treatments fail, the patient may consider surgical replacement of the affected joint.
As noted above, NSAIDs, both over-the-counter and prescription are commonly taken to manage the pain of backache, osteoarthritis, rheumatoid arthritis, headache and other painful conditions. For example, according to a study commissioned by Kitov from IMS Health, the largest vendor of U.S. physician prescribing data, between April 2015 and March 2016 there were 2,428,176 prescriptions for celecoxib dispensed in the US.
NICOX, a pharmaceutical company, has attempted to develop NAPROXCINOD®, a naproxen-based drug intended to treat pain and to act as an anti-hypertensive. From 2005 to 2010, NICOX completed three Phase III clinical trials following a significant investment. However, the results of the trials did not meet the FDA’s requirements. Therefore, in May 2010, an outside advisory committee to the FDA recommended against approving the drug. As a result of this recommendation, and its own internal review, the FDA rejected the request for NDA approval. According to an announcement by NICOX in April 2012, pursuant to an appeal filed by NICOX in July 2011, a meeting was held in April 2012 between representatives of NICOX and the FDA, in which NICOX was informed that in order to gain approval of its drug, it must file a new NDA, that would include results from additional clinical trials, for the purpose of approving a specific dosage of the drug.
In July 2015 the FDA published a safety announcement requiring labeling for prescription NSAIDs to indicate that the risk of heart attack or stroke can occur as early as the first weeks of using an NSAID and that the risk may increase with longer use of the NSAID. In effect, the current warnings indicated on the labeling, in effect since 2005, will be strengthened as a result of a review by the FDA of a variety of new safety information on prescription and over-the-counter NSAIDs, including observational studies, a large combined analysis of clinical trials, and other scientific publications. These studies were discussed at a joint meeting of the Arthritis Advisory Committee and Drug Safety and Risk Management Advisory Committee held in February 2014. As a result of its reviews of NSAIDs, the FDA has cautioned in the labeling of NSAIDs that combining an NSAID with antihypertensive drugs, including diuretics, beta blockers, ACE inhibitors, or angiotensin receptor blockers, may markedly diminish the efficacy of these antihypertensive drugs. Calcium channel blockers, such as amlodipine besylate, the anti-hypertensive component of Consensi™, were not included in this labeling requirement.
Hypertension (High Blood Pressure)
Hypertension is the most common chronic disease in the western world, affecting approximately thirty percent (30%) of the U.S. adult population, according to an article in Morbidity and Mortality Weekly Report. Untreated, hypertension can cause significant morbidity and mortality.
According to its physiological definition, “hypertension” is an excessive pressure applied by the blood on the walls of the blood vessels. The term hypertension refers to excessive arterial blood pressure, which is the pressure in the arteries that propels blood to body organs.
The blood pressure is created as a result of the contraction of the cardiac muscle propelling blood into the arteries, which possess a limited capacity to store the blood. Blood pressure is measured in units of mercury (Hg) millimeters (mm Hg). Diagnosing hypertension in adults requires at least two measures on two different occasions. There are two blood pressure values:
- Systolic pressure is the peak pressure in the arteries measured in the cardiac cycle, during the contraction of the heart’s left ventricle (systole); and
- Diastolic pressure is the lowest pressure point in the arteries measured when the heart’s left ventricle is relaxing and there is no contraction of the heart (diastole).
In the past, hypertension was generally defined as a systolic blood pressure of greater than 140 mm Hg or a diastolic blood pressure of greater than 90 mm Hg. However, as discussed below, a recently halted NIH study may result in these designated values being set lower. As a result of these data, multiple entities, including the American College of Cardiology, have recently recommended that a patient’s systolic blood pressure should be maintained at a level below 130 mm Hg, and their diastolic blood pressure maintained below 80 mm Hg.
The cause of hypertension in 95% of patients is unknown, and in these cases hypertension is defined as “essential hypertension”. However, some studies postulate that genetic factors and environmental factors are involved in the initial development of hypertension. These factors include high salt consumption, obesity, excessive alcohol consumption, and probably mental and behavioral factors, which may be caused by various circumstances, including working in certain professions. Extreme hypertension may lead to functional disorders, and worsening health, while the affected person does not necessarily feel it and/or is aware of it. Therefore, hypertension is often referred to as the “silent killer”.
The danger of hypertension is continuing damage to blood vessels in critical areas of the body, such as blood vessels in the heart, kidneys, eyes, and to the nerve tissue in the brain where any damage may cause a stroke. Moreover, damage to the blood vessels may cause blockage due to arteriosclerosis and lead to the tearing of the vessels. These complications may cause various diseases and even death.
Hypertension treatment methods focus on reducing the patient’s blood pressure to normal values, thereby preventing the occurrence of complications in the long term. Even a small increase in blood pressure may cause significant cardiovascular problems. For example, it has been shown that any increase in blood pressure above a systolic value of 115 mm Hg is associated with an increased risk of suffering a cardiovascular death. This finding has been repeatedly replicated and it is now established that there is no safe level of blood pressure increase above of the “normotensive baseline value” of approximately 120 systolic and 70 diastolic. The documentation of a danger of any increase in blood pressure above a value of 120/70 was recently documented in September of 2015 in a large NIH sponsored clinical trial which enrolled over 9000 patients age 50 and older. This study also documented that patients age 50 and older with systolic blood pressures greater than 120 had a greater rate of adverse cardiovascular events than did those whose systolic blood pressure was treated to levels below 120.
It has been recognized for many decades that hypertension requires treatment. This fact has been recently re-emphasized by a paper that reviewed 147 prior randomized studies of antihypertensive treatments. This meta-analysis study, concluded that the majority of the adult population with hypertension can be expected to benefit considerably from using anti-hypertension drugs.
Hypertension can be treated with many different classes of medications. These include diuretics, beta blockers, alpha blockers, calcium channel blockers, ACE inhibitors, angiotensin receptor antagonists and vasodilators. In general, these medications work by either relaxing blood vessels and thereby lowering the pressure in arteries, or by assisting the body in removing fluid and thereby decreasing the pressure inside of arteries.
Although drugs from each of the various classes of antihypertension medications are able to reduce blood pressure, there are marked differences in their side effects profiles. For example, the diuretics can result in kidney problems, while the beta blockers can slow the heart rate. It is therefore important for physicians carefully to select which antihypertension medications to prescribe for patients based upon the patient’s other medical problems, including what concomitant medications they are receiving.
Blood pressure can undergo significant alterations when subjects are placed on various medications. For example, according to a May 2010 FDA Joint Meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee report published by the FDA, an increase of about 3.5 mm Hg was diagnosed following the use of naproxen, while the use of Celebrex® causes an increase of about 2.5 mm Hg. In addition, in August 2011 the FDA issued a Safety Information release stating that co-administration of NSAIDs, including selective COX-2 inhibitors, with ACE inhibitors or with angiotensin II receptor antagonists, may result in deterioration of renal function, including possible acute renal failure, and that the antihypertensive effect of ACE inhibitors may be attenuated by NSAIDs. No such Safety Information release was issued with regard to calcium channel blockers, which is the anti-hypertensive used in its therapeutic candidates.
The FDA has also required warnings in the labeling of NSAIDs that adding diuretics or beta blockers to patients on NSAIDs can cause problems with the control of their blood pressure. Calcium channel blockers, such as amlodipine besylate, the anti-hypertensive component of Consensi™, were not included in this labeling requirement.
Background on Combination Products
Numerous companies worldwide have developed in recent years successful combination products comprised of a combination of two or more drugs to treat various medical conditions, where the safety and effectiveness of each of the drugs was proven separately.
Combination products manufactured and sold, which are similar to its therapeutic candidate, include:
- Vimovo®, which was developed by Aralez Pharmaceuticals Inc. (originally Pozen Inc.) and was approved by the FDA in May 2010. Vimovo® is a combination of naproxen and esomeprazole magnesium, marketed by AstraZeneca PLC worldwide (except in the U.S.) and by Horizon Pharma in the U.S., and is designed for treating both pain and preventing gastric ulcer. Vimovo’s® net sales in the U.S. reached $121 million in 2016, compared to net sales of $163 million in 2014.
- Caduet®, a combination of Lipitor® and amlodipine, was originally developed and manufactured by Pfizer and is designated for treating both cholesterol and hypertension, with global sales of $193 million in 2015.
- Janumet®, a combination of metformin and sitagliptin, manufactured by Merck & Co. Inc. and designated to treat diabetes, with sales of $2,201 million in 2016.
- Treximet®, a combination of naproxen and sumatriptan, was originally developed by Aralez Pharmaceuticals Inc. (originally Pozen Inc.) and marketed by Pernix Inc., and designed for relief of headache, pain, and other migraine symptoms, with U.S sales of $66.9 million in 2016.
Combination drugs may provide improved medical treatment of patients diagnosed as suffering from two or more different diseases and also may provide convenience to patients by using a single drug instead of multiple drugs. In addition, combination drugs have significant commercial advantages deriving from maintaining and even increasing the market share of the active ingredients after their patents expire by extending the life span of the patents for the active ingredients through the use of combination drugs.
Therapeutic Candidate
Studies estimate that approximately 13.5 million patients in the U.S. alone may suffer concurrently from hypertension and chronic osteoarthritis pain in the joints, according to data published by the CDC. Kitov Pharamceuticals is developing Consensi™ based on the generic drugs celecoxib and amlodipine besylate. Celecoxib is the active ingredient in the branded drug Celebrex®, a known and approved-for-use drug designed primarily to relieve pain caused by osteoarthritis. The company's combination is designed simultaneously to relieve pain caused by osteoarthritis and treat hypertension, which is one of the side effects of using NSAIDs for treating pain caused by osteoarthritis. The company's strategy in developing Consensi™ is based on its belief that the added anti-hypertensive drug will decrease the side effect of increased hypertension typically caused by the use of NSAIDs alone.
To date, no combination drug exists that offers the combined treatment of pain caused by osteoarthritis and hypertension. The company therefore believe that Consensi™ potentially holds significant advantages over the currently available drugs in the market, due to the fact that the drug treatment of osteoarthritis together with hypertension eases the burden of the treatment process for patients by providing the ability to use one drug instead of multiple drugs concurrently, thereby increasing the patients’ ease of compliance with the required treatment.
Consensi™ is a fixed dosage combination product based on two known active ingredients (celecoxib and amlodipine besylate), the effectiveness and safety of which has been separately proven for each, and which is intended to enable the concurrent treatment of pain caused by osteoarthritis, and hypertension.
On November 7, 2013, the company filed with the FDA the final statistical plan for the Phase III clinical trial protocol for Consensi™ as part of the SPA procedures. On February 20, 2014, the FDA replied and indicated that the proposed data analysis of the trial’s results that the company submitted to the FDA provides a suitable solution to achieve the primary endpoint of the Phase III clinical trial and to support the final request for approval, which will be submitted. As a result of the SPA process, the FDA approved the Phase III trial design for its clinical trial, and cleared its clinical trial to begin, and on June 18, 2014, the company commenced the clinical trial, as described below under “Research and Development”. The Phase III clinical trial was performed using the Adaptive Trial Design method, or ATD, in accordance with the SPA. Based on the ATD format, in the first stage of the trial 150 patients were to be recruited. Then, the decision as to whether or not to add additional patients was to be based upon a statistical calculation of the preliminary data performed by an independent statistician appointed by the Company in accordance with the protocol of the clinical trial, and in accordance with the FDA’s requested and approved method as part of the SPA, as agreed upon between the FDA and the Company. Statistical analysis of the preliminary data collected in the Phase III clinical trial was performed by the independent statisticians in accordance with the clinical trial protocol, and showed that the study met the pre-specified criteria the FDA required for stopping patient enrollment and thus proceeding to the completion of the final statistical analyses. The final statistical analyses of the data demonstrated that the Phase III study of Consensi™ met its FDA approved primary efficacy endpoint with statistical significance based upon the efficacy endpoint of less than 0.001.
Additional data from the Phase III clinical trial of Consensi™ suggested beneficial effects on renal (kidney) function, as compared to negative effects on renal function caused by other NSAIDS. In addition, Kitov Pharamceuticals has completed a Phase III/IV clinical trial designed to validate and better quantify these potential beneficial renal effects. The trial was designed to explain the synergistic antihypertensive effect, where the reduction in diastolic blood pressure demonstrated with the components of Consensi™ was greater than that observed with amlodipine besylate alone at certain times of the day. Accordingly, the company conducted a double blind, placebo controlled, clinical trial intended statistically to demonstrate Consensi™’s effects on renal and vascular function, while providing it with data with respect to Consensi™ in addition to the data of the Phase III clinical trial, by utilizing a primary efficacy end-point in the renal function clinical trial comparable to that of the Phase III clinical trial. The primary efficacy endpoint of the trial was to show that Consensi™ lowers daytime systolic blood pressure by at least 50% of the reduction in blood pressure achieved in patients treated with amlodipine besylate only. Secondary endpoints included various parameters of renal function. In October 2017, the company announced that Phase III/IV renal function clinical trial, successfully met its primary efficacy endpoint. Data from the trial demonstrated that Consensi™ lowered systolic blood pressure a comparable amount to amlodipine besylate, thus meeting the trial’s primary efficacy endpoint of achieving at least 50% of the amlodipine reduction (p=0.019). The study also demonstrated that treatment with Consensi™ led to a statistically significant reduction of serum creatinine, a marker of renal function, from its baseline value (p=0.0005). In contrast, neither amlodipine besylate nor placebo lowered creatinine to a statistically significant level. When comparing the effect of Consensi™ to amlodipine besylate in lowering creatinine, it was found that Consensi™ enhanced the creatinine reduction by an average of 102% over that achieved with amlodipine besylate alone, although there was a slight, but statistically insignificant, increase in the rate of edema in the Consensi™ treatment arm. Although the Phase III/IV renal function clinical trial was not required as part of the initial Consensi™ NDA submission to the FDA, the company delivered the initial study results data to the FDA shortly following completion of the study, and anticipate that the company will submit the completed Phase III/IV renal function clinical study report to the FDA within six to eight weeks of this Annual Report on Form 20-F.
Below is a summary of its projected timeline for the development of Consensi™:
| Current Status | 2018 | 2019 |
|---|---|---|
| FDA Approved SPA. | Anticipated FDA approval for marketing | Continuation of our business development activity. |
| Phase III clinical trial completed. | Continuation of our business development activity. | |
| CMC, including stability testing, completed. | ||
| PK studies completed. | ||
| NDA filed by FDA and PDUFA date set by FDA for May 31, 2108 | ||
| FDA review of NDA and Kitov follow-up submissions. | . | . |
Consensi™ is based on two generic drugs (amlodipine besylate and celecoxib). Until December 2015 celecoxib was protected by patents held by Pfizer Inc. (Celebrex®). The USPTO granted Pfizer a “reissue patent” covering methods of treating osteoarthritis and other approved conditions with celecoxib, the active ingredient in Celebrex®. The reissued patent extended U.S. patent protection for Celebrex® from May 30, 2014 to Dec. 2, 2015.
The company submitted the New Drug Application (NDA) for marketing approval of Consensi™ to the U.S. Food and Drug Administration (FDA) in July 2017, and on October 2, 2017 the company announced that the FDA filed the NDA, thereby granting a full review. In connection with its determination that its application is sufficiently complete to permit a substantive review, the FDA, under the Prescription Drug User Fee Act (PDUFA), has set a target date of May 31, 2018 to complete its review. Although its Phase III/IV renal function clinical trial was not required as part of the initial Consensi™ NDA submission to the FDA, the company delivered the initial study results data to the FDA shortly following completion of the study, and the company expect to submit the completed Phase III/IV renal function clinical study report to FDA within six to eight weeks of this Annual Report on Form 20-F. The FDA has indicated to it that a submission of this report at such time could possibly result in the extension of the PDUFA date by up to an additional 90 days, but have not definitely indicated that they would extend the PDUFA date. The company's management is of the view that the submission of the Phase III/IV renal function clinical study report to the FDA has the potential to strengthen the drug’s labeling and support future marketing of Consensi™, and that the potential labeling and marketing benefits that could be derived from submission of the Phase III/IV renal function clinical study report to the FDA are substantially more important to Consensi™’s commercial prospects than a possible short-term delay in obtaining marketing approval. The results of the Phase III/IV renal function clinical trial validated the beneficial blood pressure reducing effects demonstrated by Consensi™ in its initial efficacy study in an additional clinical population beyond that included in the original Phase III trial. The Phase III/IV study evaluated patients with chronic hypertension, while the initial Phase III clinical trial included only newly diagnosed hypertensive patients. These results indicate a potentially expanded patient target market for Consensi™. Additionally, the results of the Phase III/IV study also demonstrated that when patients with chronic hypertension are treated with Consensi™, their renal function, as assayed by serum creatinine, improves over time. As renal toxicity is a significant issue for patients being treated with the entire class of NSAID drugs, this clinical finding could also differentiate Consensi™ from other NSAIDs.
During the NDA review period, as is common for NDA reviews, Kitov Pharamceuticals has been responding to FDA information requests on an ongoing basis. In light of such information requests, the company cannot make assurances that the FDA will not require it to submit additional data, or complete additional studies in connection with Consensi™, prior to considering the issuance of marketing approval for Consensi™. For example, as part of the NDA review process the FDA has asked it to provide additional data in connection with the chemistry of the over-encapsulation of the pills given to the patients in the Phase III clinical trial. Such requests and other possible requests for additional data or studies, as well as the possibility that the FDA may consider the submission of the Phase III/IV renal function trial clinical study report to be a major amendment to the NDA which would allow the FDA to extend the PDUFA date by up to 90 days, may delay the FDA approval of its NDA, and otherwise impact the NDA submission for Consensi™ in a manner not currently known to it. In any event, however, the company still anticipate receiving approval from the FDA to market Consensi™ later this year. As a result of this timing and because Consensi™ combines the treatment of osteoarthritis by celecoxib with amlodipine besylate, which treats the side effect of hypertension, and that when patients with chronic hypertension are treated with the two components of Consensi™, their renal function, improves over time, and as renal toxicity is a significant issue for patients being treated with the entire class of NSAID drugs, the company believe that Consensi™ may be an attractive alternative to the marketed generic versions of Celebrex® and other NSAID drugs
Research and Development – KIT 302
Consensi™ is a fixed dose combination drug comprised of known and approved-for-use components, the combination of which is intended simultaneously to treat the pain caused by osteoarthritis and reduce blood pressure, thereby offsetting a side effect caused by the use of NSAIDs for osteoarthritis. Following discussions with the FDA, the FDA approved a development design in accordance with the 505(b)(2) NDA track. The FDA did not require it to perform pre-clinical trials in animal models, and therefore Kitov Pharamceuticals is required only to conduct a single Phase III clinical trial and standard pharmacokinetic trials and bioequivalence trials.
For the development of Consensi™, the company performed a double blind, placebo controlled, Phase III clinical trial for testing the decrease of hypertension in patients receiving the two components of its Consensi™ therapeutic candidate. This trial was performed in the U.K. in four groups of twenty-six (26) to forty-nine (49) patients (a total of 152 patients), with each patient treated over a total period of two weeks. Group One was treated with the two components of Consensi™ (celecoxib and amlodipine besylate), Group Two was treated with a standard drug available in the market for treating hypertension (amlodipine besylate, one of the components of Consensi™), Group Three was treated with celecoxib only, and Group Four received a double placebo. The trial began in June 2014, and the final patient completed the study in November 2015.
The purpose of the trial was to show that a combination of the two components of Consensi™, as demonstrated in Group One, lowered blood pressure by at least 50% as compared to the reduction in blood pressure in patients in Group Two (treatment with amlodipine besylate only). Kitov Pharamceuticals was not required by FDA to demonstrate or measure efficacy in treatment of pain caused by osteoarthritis. Group Three and Group Four were included for control purposes and would not be considered in evaluating the primary efficacy endpoint. The trial was conducted with off-the-shelf drugs. Consensi™, its combination drug, was being developed in parallel by Dexcel Ltd., or Dexcel. The trial was conducted with only one dosage of amlodipine besylate (10 mg), although based on the SPA signed with the FDA, the company expect that having conducted a trial with this dosage only, will be sufficient to seek marketing approval from the FDA for three dosages (10mg, 5 mg, and 2.5 mg), each combined with 200 mg of celecoxib. The company announced the top line trial results in December 2015, showing that the company successfully met the primary efficacy endpoint of the trial protocol as approved by the FDA. Data from the trial further revealed that Consensi™ tended to reduce blood pressure more than the widely used hypertension drug amlodipine besylate alone.
The trial’s interim results demonstrated that the number of 152 patients treated was adequate to provide statistical validity and therefore, the results were final. These final results showed that in patients treated with amlodipine besylate only, there was a mean reduction in daytime systolic blood pressure of 8.8 mm Hg. In patients treated with Consensi™’s two components, there was a mean reduction in daytime systolic blood pressure of 10.6 mm Hg. Therefore, the primary efficacy endpoint of the study has been successfully achieved with a p value of 0.001.
Additional data from the trial results showed that favorable blood pressure effects of Consensi™’s two components were present in all blood pressure variables measured in the study. The data indicated that the blood pressure reduction synergy seen with combining celecoxib and amlodipine, is seen not only in the study’s primary efficacy endpoint of daytime systolic blood pressure, but was also seen from daytime diastolic blood pressure measurements and in all other blood pressure variables. After two weeks of treatment, the reduction with daytime diastolic blood pressure measurements with amlodipine alone was 5.5 mm Hg, while for patients treated with Consensi™’s components the reduction was 7.6 mm Hg. For nighttime systolic blood pressure after two weeks of treatment, the reduction with amlodipine therapy alone was 6.3 mm Hg, while for patients treated with Consensi™’s components the reduction was 10.7 mm Hg. For nighttime diastolic blood pressure after two weeks of treatment, the reduction with amlodipine besylate alone was 3.1 mm Hg, while for patients treated with Consensi™’s components the reduction was 7.2 mm Hg. Thus, the synergy in blood pressure reduction demonstrated with Consensi™’s two components was present at all times of day and with both blood pressure measures. Although celecoxib when combined with amlodipine appears to have a synergistic effect in lowering blood pressure, it appears to have the opposite effect when administered by itself.
In July 2017 the company submitted the New Drug Application (NDA) for marketing approval of Consensi™ to the U.S. Food and Drug Administration (FDA), and on October 2, 2017 the company announced that the FDA filed the NDA, thereby granting a full review. In connection with its determination that its application is sufficiently complete to permit a substantive review, the FDA, under the Prescription Drug User Fee Act (PDUFA), has set a target date of May 31, 2018 to complete its review.
In addition, in connection with its Development Services Agreement with Dexcel, pursuant to which Dexcel developed the formulation for Consensi™ and performed the subsequent stability testing and manufacturing scale-up in quantities adequate for submission of an NDA to the FDA, Dexcel performed conclusive pharmacokinetic (PK) bioequivalence (BE) studies. The objective of these studies was to check the pharmacokinetics of the combination drug in order to show that the blood levels achieved with its combination are equivalent to those obtained with the individual components.
The PK Studies were conducted under both fed and fasted conditions, using the 10 mg amlodipine component, and compared the PK of Consensi™ containing the higher dosage (10 mg) of amlodipine to off-the-shelf branded 200 mg celecoxib capsules and 10 mg amlodipine tablets. The results demonstrated that for both the Cmax (the maximum blood level achieved) and AUC (the area under the concentration-time curve for drug levels), the 90% confidence intervals for both the amlodipine and celecoxib components of Consensi™ were documented to be between 80% and 125% of the values obtained with the off-the-shelf drugs, thus meeting the FDA’s standard for establishing bioequivalence. A similar PK bioequivalence study for Consensi™, containing a lower dosage (2.5 mg) of amlodipine, was also completed, and showed similar bioequivalence results to those found in the Final PK Study.
The Phase III clinical trial for Consensi™ was conducted in medical centers in the United Kingdom on the basis of approvals received from the British Regulatory Authority (MHRA) and the U.K. ethics committees.
Additional data from the Phase III clinical trial of Consensi™ also suggested beneficial effects on renal (kidney) function, as compared to negative effects on renal function caused by other NSAIDS. Greater reduction in plasma levels of creatinine was observed in patients in the Consensi™’s two components arm (-3.22 umol/L) compared to creatinine reduction observed in patients in the amlodipine arm (-2.55 umol/L), suggesting better renal function. In addition, peripheral edema, a known side effect of calcium channel blockers such as amlodipine, was reported in 15.6% of patients receiving amlodipine alone, but in only 8.2% of patients receiving Consensi™’s two components, suggesting that Consensi™ may protect against the amlodipine side effect of causing fluid retention by the kidneys. It is recognized that such an effect could explain at least in part, the synergistic blood pressure reducing effect of Consensi™ over therapy with amlodipine alone.
Although not intended as part of the information to be included in its new drug application that the company submitted for the marketing clearance by the FDA of Consensi™, the company completed conducting a Phase III/IV clinical trial designed to validate and better quantify these potential beneficial renal effects. The trial was designed to explain the synergistic antihypertensive effect, where the reduction in blood pressure demonstrated with Consensi™’s two components was greater than that observed with amlodipine alone. Accordingly, the company conducted a double blind, placebo controlled, clinical trial intended statistically to demonstrate Consensi™’s effects on renal and vascular function, while providing it with data with respect to Consensi™ in addition to the data of the Phase III clinical trial, by utilizing a primary efficacy end-point in the renal function clinical trial comparable to that of the Phase III clinical trial. The trial was performed in the U.K. in three groups of 8 to 49 patients (and a total of 104 patients), with each patient treated over a total period of two weeks. Group One received a placebo, Group Two was treated with a standard drug available in the market for treating hypertension (amlodipine besylate, one of the components of Consensi™), and Group Three was treated with the two components of Consensi™ (celecoxib and amlodipine besylate). The primary efficacy endpoint of the trial was to show that Consensi™ lowers daytime systolic blood pressure by at least 50% of the reduction in blood pressure achieved in patients treated with amlodipine besylate only. Secondary endpoints included various parameters of renal function.
The Phase III/IV renal function clinical trial for Consensi™ was conducted in medical centers in the United Kingdom on the basis of the approval of the British Regulatory Authority (MHRA), as well as the approvals of the relevant U.K. ethics committees.
The company estimate that the total cost of all service providers with respect to the Phase III/IV renal function clinical trial, will amount to approximately $1.8 million.
In October 2017, the company announced that Phase III/IV renal function clinical trial, successfully met its primary efficacy endpoint. Data from the trial demonstrated that Consensi’s™ two components lowered systolic blood pressure a comparable amount to amlodipine besylate, thus meeting the trial’s primary efficacy endpoint of achieving at least 50% of the amlodipine reduction (p=0.019). The study also demonstrated that treatment with Consensi’s™ two components led to a statistically significant reduction of serum creatinine, a marker of renal function, from its baseline value (p=0.0005). In contrast, neither amlodipine besylate nor placebo lowered creatinine to a statistically significant level. When comparing the effect of Consensi’s™ two components to amlodipine besylate in lowering creatinine, it was found that Consensi’s™ two components enhanced the creatinine reduction by an average of 102% over that achieved with amlodipine besylate alone, although there was a slight, but statistically insignificant, increase in the rate of edema in the treatment arm containing Consensi’s™ two components. Although the Phase III/IV renal function clinical trial was not required as part of the initial Consensi™ NDA submission to the FDA, the company delivered the initial study results data to the FDA shortly following completion of the study, and the company expect to submit the completed Phase III/IV renal function clinical study report to FDA within six to eight weeks of this Annual Report on Form 20-F. The FDA has indicated to it that a submission of this report at such time could possibly result in the extension of the PDUFA date by up to an additional 90 days, but have not definitely indicated that they would extend the PDUFA date. The company's management is of the view that the submission of the Phase III/IV renal function clinical study report to the FDA has the potential to strengthen the drug’s labeling and support future marketing of Consensi™, and that the potential labeling and marketing benefits that could be derived from submission of the Phase III/IV renal function clinical study report to the FDA are substantially more important to Consensi™’s commercial prospects than a possible short-term delay in obtaining marketing approval. The results of the Phase III/IV renal function clinical trial validated the beneficial blood pressure reducing effects demonstrated by Consensi™ in its initial efficacy study in an additional clinical population beyond that included in the original Phase III trial. The Phase III/IV study evaluated patients with chronic hypertension, while the initial Phase III clinical trial included only newly diagnosed hypertensive patients. These results indicate a potentially expanded patient target market for Consensi™. Additionally, the results of the Phase III/IV study also demonstrated that when patients with chronic hypertension are treated with Consensi™, their renal function, as assayed by serum creatinine, improves over time. As renal toxicity is a significant issue for patients being treated with the entire class of NSAID drugs, this clinical finding could also differentiate Consensi™ from other NSAIDs.
During the NDA review period, as is common for NDA reviews, Kitov Pharamceuticals has been responding to FDA information requests on an ongoing basis. As part of the NDA review process the FDA has asked it to provide additional data in connection with the chemistry of the over-encapsulation of the pills given to the patients in the Phase III clinical trial, and Kitov Pharamceuticals is preparing the data requested for submission to the FDA. However, the company cannot make assurances that the FDA might not require it to submit additional data, or complete additional studies in connection with Consensi™, prior to considering the issuance of marketing approval for Consensi™. Such requests and other possible requests for additional data or studies, as well as the possibility that the FDA may consider the submission of the Phase III/IV renal clinical study report to be a major amendment to the NDA which would allow the FDA to extend the PDUFA date by up to 90 days, may delay the FDA approval of its NDA, and otherwise impact the NDA submission for Consensi™ in a manner not currently known to it.
Competitive Treatments for Pain Caused by Osteoarthritis
The competition for Consensi™ is expected to come from the oral anti-arthritic market, or more specifically the traditional non-selective NSAIDs (such as naproxen and ibuprofen), traditional NSAID/gastroprotective agent combination products or combination product packages (such as Vimovo®, Arthrotec®, Prevacid® and NapraPAC™) and the most common COX-2 inhibitor in the U.S. market, Celebrex® (including generic versions of Celebrex®). In 2017 global sales of Celebrex® (not including generic versions of Celebrex®) were $775 million out of which $164 million were recorded in the US, $28 million in Europe, and $583 million in the rest of the world.
Due to the voluntary withdrawal of Vioxx® by Merck & Co. in September 2004, the FDA ordered the withdrawal of Bextra® by Pfizer and issued a Public Health Advisory in April 2005, requiring manufacturers of all prescription products containing NSAIDs to provide warnings regarding potential adverse cardiovascular events as well as life-threatening gastrointestinal events associated with the use of NSAIDs. Moreover, subsequent to an FDA advisory committee meeting in February 2005 that addressed the safety of NSAIDs, and, in particular, the cardiovascular risks of COX-2 selective NSAIDs, the FDA has indicated that long-term studies evaluating cardiovascular risk will be required to approve new NSAID products that may be used on an intermittent or chronic basis. The company believe that Consensi™ has a competitive advantage over other drugs in the market because, as a COX-2 inhibitor, it has limited gastrointestinal side effects, and due to the addition of amlodipine besylate it is designed to address existing hypertension and the cardiovascular side effects of NSAIDs.
License Agreement for Territory of South Korea
On March 8, 2017, the company announced that the Company signed a definitive License Agreement Consensi™, for the territory of South Korea, with Kuhnil Pharmaceutical Co., Ltd. (“Kuhnil”), a South Korean pharmaceutical company. Upon receipt of marketing authorization in South Korea, Kuhnil will have the exclusive right and license to manufacture, distribute and sell Consensi™ in South Korea. Kuhnil will be responsible for seeking regulatory approval for Consensi™ in South Korea. Under the terms of the license agreement, Kitov is entitled to receive milestone payments upon achievement of certain predefined regulatory milestones, as well as double digit royalties in a range between ten and twenty percent of net sales. The initial term of the definitive agreement with Kuhnil is for ten years from the date of first commercial sale and shall automatically renew for an additional one-year term. Commercial launch in South Korea is estimated to take place in 2019.
In addition to its internal business development team, Kitov Pharamceuticals has engaged consultants who are assisting it with finding other potential collaboration partners for Consensi™ in various markets world-wide, with a current emphasis on the North American and Asian markets, particularly China.
NT219
In January 2017, the company acquired a majority of the shares in TyrNovo which is developing the NT219 therapeutic candidate. NT219 is a small molecule that presents what the company believe is a new concept in cancer therapy by promoting the degradation of two oncology-related checkpoints, Insulin Receptor Substrates (IRS) 1 and 2 as well as the inhibition of Signal Transducer and Activator of Transcription 3 (STAT3). In pre-clinical trials in PDX models, NT219, in combination with several approved cancer drugs, displayed potent anti-tumor effects and increased survival in various cancers by preventing the tumors from developing resistance to the approved drug treatments and re-sensitizing tumors to the approved drugs even after resistance is acquired. The NT219 technology has been tested in a number of Patient-Derived Xenograft (PDX) models where human primary cancer cells or biopsies are taken and transplanted into mice and then used to test various cancer drugs.
Below is a summary of its current projected timeline for the development of NT219:
| Current Status | 2018 | 2019 |
|---|---|---|
| Efficacy demonstrated in | Complete GLP manufacturing of drug for pre-clinical and toxicology studies. | Submit IND. |
| various PDX models. Additional experiments ongoing. | Conduct cGMP manufacturing of drug for IND and clinical trials. | Initiate clinical trials. |
| Academic collaborations established. | Complete preclinical studies. | |
| Held pre-IND meeting with FDA | Conduct GLP toxicology studies. | |
| CMC work and toxicology studies initiated. | . | . |
Background on Cancer Drug Resistance
The following are high-level summaries of the therapeutic areas Kitov Pharamceuticals is currently investigating for NT219:
Solid malignancies (e.g., pancreatic, colon and non-small cell lung cancer). According to the Journal of Oncology Practice, in 2020 roughly 1 in every 19 people worldwide will either be diagnosed with a solid tumor or be a cancer survivor. According to the American Cancer Society, lung cancers are the most common cause of cancer deaths worldwide, while pancreatic cancers are the third most common cause of cancer-related deaths in the United States. Pancreatic, colon and non-small cell lung malignancies have high mortality rates and poor five-year survival prognosis. Novel, emerging therapeutic approaches for targeting solid tumors are being developed and tested.
Tumor Resistance to Cancer Therapies. Resistance to chemotherapy and to targeted therapies is a major problem facing current cancer research. The mechanisms of resistance to ‘classical’ cytotoxic chemotherapeutics and to therapies that are designed to be selective for specific target proteins share many features, such as alterations in the drug target, activation of pro-survival pathways and ineffective induction of cell death.
Recent evidence suggests that among other mechanisms of resistance, inhibition of central oncological target kinases such as EGFR, MEK and mutated-BRAF could trigger feedback activation of STAT3 and IRS-to-PI3K/AKT, major survival pathways that bypass (prevent) the anti-cancer effects of various drugs.
IRS. Insulin Receptor Substrate (IRS) is a junction protein that mediates various mitogenic and anti-apoptotic signals mainly from Insulin-like Growth Factor-1 Receptor (IGF1R) and Insulin Receptor (IR), but also from other oncogenes such as v-Src and ALK-fusion proteins. IRS expression is often increased in human tumors, such as prostate, pancreatic, liver, renal and ovarian cancer. Resistance to several anti-cancer therapies (e.g. inhibitors of EGFR, MEK, mutated-BRAF, mTOR, as well as chemotherapy) may be mediated by IRS up-regulation, as demonstrated in peer reviewed research articles which have been published in scientific journals.
STAT3. Signal Transducer and Activator of Transcription 3 (STAT3) plays crucial roles in several cellular processes such as cell proliferation and survival, and has been found to be aberrantly activated in many cancers (such as NSCLC, pancreatic cancer and many others). Much research has explored the leading mechanisms for regulating the STAT3 pathway and its role in promoting tumorigenesis. Evidence suggests that feedback activation of STAT3 plays a prominent role in mediating drug resistance to a broad spectrum of targeted cancer therapies and chemotherapies (such as inhibitors of EGFR, MEK, ALK, as well as 5FU, Oxaliplatin and SN-38).
Mechanism of Action – NT219
The NT219 therapeutic candidate is a small molecule that the company believe presents a new concept in cancer therapy, acting as a dual inhibitor of IRS and STAT3, both of which play major roles in cancer and drug resistance. While targeted anti-cancer drugs inhibit the “ON” signal, NT219 activates the “OFF” switch, leading to the degradation of IRS-1 and IRS-2 and extensively blocking major oncogenic pathways.
IRS down-regulation can be mediated by several oncogenic pathways (EGFR, MAPK, mTOR, etc.). Blockade of these pathways by various drugs, could inhibit serine phosphorylation of IRS, leading to the activation of IRS to AKT survival bypass. Therefore, elimination of IRS1/2 by NT219 potentially could prevent resistance and prolong the tumor’s response to various targeted drugs, as depicted below:
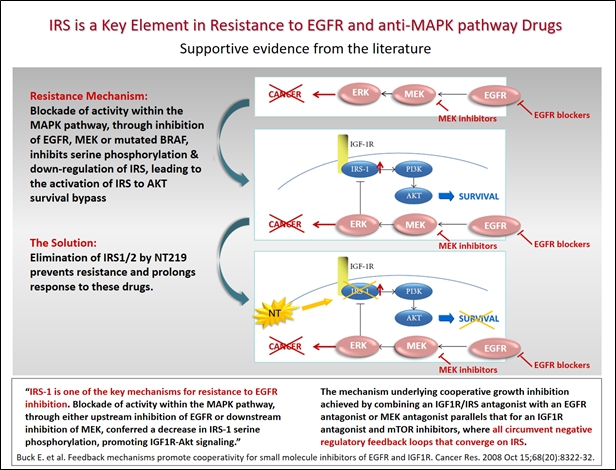
There have been reports in peer reviewed academic literature describing the involvement of Insulin-like Growth Factor-1 Receptor (IGF1R) up-regulation in drug-resistance. In these cases, blockage of IGF1R direct substrates, IRS1/2, by NT219 could potentially overcome drug resistance.
The same principal is true for STAT3. Feedback activation of STAT3 is a common cause of resistance to many targeted cancer therapies (such as the inhibitors of EGFR, MEK, HER2) and chemotherapies. Combining these cancer therapies with NT219, which disrupt this feedback mechanism, could potentially enhance cell death and delay resistance, suggesting a co-treatment strategy that may be broadly effective in oncogene-addicted tumors.
Elimination of IRS proteins and blockage of STAT3 by NT219 could potentially prevent resistance to multiple anti-cancer drugs, extend the duration of effective drug treatment, and restore drug sensitivity in resistant tumors.
Preclinical results - NT219
In pre-clinical trials, NT219, in combination with several approved cancer drugs, displayed potent anti-tumor effects and increased survival in various cancers by preventing the tumors from developing drug resistance and restoring sensitivity to the drugs after resistance is acquired. The NT219 molecule has been tested in a number Patient-Derived Xenograft (PDX) models where human primary cancer cells or biopsies are taken and transplanted into mice and then used to test various cancer drugs. NT219 has shown efficacy in various PDX models originated from non-small cell lung cancer (NSCLC), sarcoma, melanoma, pancreatic, head & neck and colon cancers.
Efficacy of NT219 was demonstrated in combination with three major families of oncology drugs:
- Antibodies such as the anti-epidermal growth factor receptor (EGFR) antibody (Erbitux®) and the immuno-oncology anti-PD1 antibody (Keytruda®);
- Kinase Inhibitors such as blockers of EGFR (Tagrisso®, Tarceva®), MEK (Mekinist®), Mutated BRAF (Zelboraf®), and mTOR (Afinitor®); and
- Chemotherapy agents such as Gemzar®, 5FU, and Oxaliplatin.
Below are two examples of results obtained with NT219 in PDX models. In the head and neck cancer model treatment with NT219 in combination with Elrotinib (trade name Tarceva®, an anti EGFR drug approved for various oncology indications; left panel) resulted in overcoming drug resistance and lower volume of the tumor, compared to the treatment arm with Erlotinib alone. Similar results are depicted on the right panel where NT219 was tested in a pancreatic cancer PDX model where combination of NT219 with gemcitabine (a chemotherapy agent approved for pancreatic cancer) resulted in decreased tumor volume compared to those obtained with gemcitabine alone.
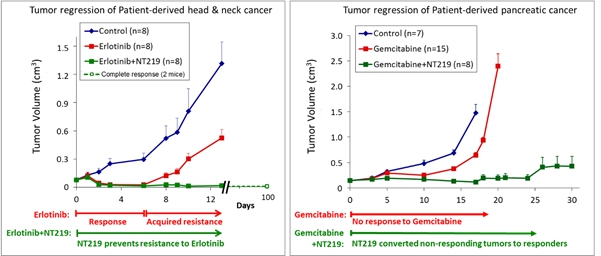
The above are examples only, and do not serve as indication of the nature of the cancers that the company expect NT219 to be tested on, or to eventually assist in treating. Based on its pre-clinical work and pre-IND correspondence with the FDA, Kitov Pharamceuticals is considering initiating NT219 clinical studies in combination with gemcitabine (GemzarTM) for the treatment of pancreatic cancer and/or in combination with osimertinib (TagrissoTM) for the treatment of non-small cell lung cancer (NSCLC).
In November 2017 the company announced that TyrNovo received the FDA’s response to the NT219’s pre-IND submission package. The FDA has agreed to the proposed Chemistry Manufacturing and Controls (CMC), preclinical, and clinical development plans for NT219. For the clinical development plan, the FDA agreed with TyrNovo’s proposed development plan to test NT219 in combination with gemcitabine for the treatment of advanced pancreatic cancer. The FDA further agreed that the initial clinical trial with NT219 will be a Phase I/II clinical trial, and that “the overall design of proposed first-in-human trial appears reasonable”. The FDA further agreed that one-month animal toxicology studies for NT219 alone would be sufficient to support the IND and that no toxicology studies of NT219 together with gemcitabine would be necessary. Kitov Pharamceuticals is moving forward with these development plans and based on its current development plans, the company expect to submit the IND during the first half of 2019. Initially the company will test NT219 in combination with gemcitabine on advanced pancreatic cancer patients, based on its consistent encouraging results in preclinical PDX models. The company's long-term strategy is to develop NT219 in combination with other oncology drugs and for additional oncology indications, by itself or in collaboration with potential strategic partners.
Competitive Oncology Drugs in Development that Target IRS1/2 or STAT3
While Kitov Pharamceuticals is not familiar with other molecules which act as dual inhibitors of both IRS1/2 and STAT3, or lead to degradation of IRS1/2, and which are in late stage of development, there are several therapeutic candidates in development which target either upstream target of IRS1/2 as Insulin Like Growth Factor 1 Receptor (IGF1R), such as Dalotuzumab (a recombinant humanized monoclonal antibody, developed by Merck & Co for metastatic breast cancer), or target STAT3 such as Napabucasin (which is developed by Boston Biomedical and designed to inhibit cancer stem cell pathways), which are currently at Phase III clinical trial state for metastatic pancreatic and colon cancers. There are also other therapeutic candidates that target these pathways, which are mostly in early stage of development.
While, based on its results in preclinical PDX models, Kitov Pharamceuticals is considering to initially test NT219 in combination with gemcitabine on advanced pancreatic cancer patients, its long-term strategy is to also develop NT219 for use in combination with other oncology drugs and for additional oncology indications in collaboration with potential strategic partners. Since Kitov Pharamceuticals has not yet finalized its complete selection of the clinical indications, and the final target drugs have not been chosen to be administrated in combination with NT219, Kitov Pharamceuticals is at this stage unable to determine the future competitive landscape of this therapeutic candidate.
Intellectual Property
Patents, trademarks and licenses and market exclusivity
The company's policy is to seek to protect its proprietary position by, among other methods, filing U.S. and foreign patent applications related to its proprietary technology, inventions and improvements that are important to the development of its business. The company also rely on its trade secrets, know-how and continuing technological innovation to develop and maintain its proprietary position. The company vigorously defend its intellectual property to preserve its rights and gain the benefit of its technological investments. The company's business is not dependent, however, upon any single patent, trademark or contract. See “Item 3. Key Information – D. Risk Factors – Risks Related to Intellectual Property”.
Kitov
Kitov Pharma owns two U.S. patents and the company expect to be pursuing additional international patent applications relating to its lead drug candidate, Consensi™. The following is a brief description of Kitov Pharma’s patent and trademark-related intellectual property:
On August 10, 2016, the company announced that the United States Patent and Trademark Office (USPTO) issued patent #9,408,837 covering Consensi™. The patent, entitled “Ameliorating Drug-Induced Elevations In Blood Pressure By Adjunctive Use Of Antihypertensive Drugs,” was issued on August 9, 2016 and is expected to have a term that can extend to February 28, 2030. The patent includes claims covering methods of ameliorating celecoxib-induced elevation of blood pressure by administering celecoxib and amlodipine separately or in combination.
On May 30, 2017 the USPTO issued patent #9,662,315 covering an oral dosage composition which includes both celecoxib and amlodipine. This patent was a divisional of the ’837 patent and its term will run concurrently with that patent.
On July 6, 2017, the company filed a U.S. provisional application in partnership with Dexcel, Ltd. which is related to pharmaceutical formulations of celecoxib and amlodipine and methods of preparing the same. The company will be filing an international application based on the U.S. provisional application by July 6, 2018, and will proceed with filings in various countries and jurisdictions based on the international application in 2019.
In December 2017 the company announced that the FDA had granted permission to use the trademark Consensi™, subject to receipt of marketing approval from the FDA for the therapeutic candidate. In January 2018 the company received from the USPTO a notice of allowance for the trademark Consensi™.
TyrNovo
TyrNovo’s patent and patent application portfolio includes five patent families, covering compounds that modulate protein kinase signaling and their use in treatment of protein kinase related disorders, including cancer and neurodegenerative disorders.
- Patent Family 1 was filed on December 4, 2007 (PCT filing date). The priority date is December 4, 2006. This family is directed to compounds modulating the insulin like growth factor receptor signaling and methods of using these compounds as chemotherapeutic agents for the treatment of protein kinase related disorders, in particular cancer. National phase counterparts exist in Europe (EP 2125712) and the United States (US 8,058,309), both of which are now granted. EP 2125712 has a maximum term of December 4, 2027, while US 8,058,309 has a maximum term of April 2, 2028 (each not including any available patent term extension (PTE)). The European patent was validated in France, Germany, Switzerland and the United Kingdom.
- Patent Family 2 was filed on June 7, 2009 (PCT filing date). The priority date is June 5, 2008. This family is also directed to compounds modulating the insulin like growth factor receptor signaling, and methods of using these compounds as chemotherapeutic agents for the treatment of protein kinase related disorders, in particular cancer. This patent family specifically discloses and claims NT-219. National phase counterparts exist in Europe (EP 2285774), the United States (US 8,637,575) and Israel (IL 209638), all of which are now granted. EP 2285774 and IL 209638 have a maximum term of June 7, 2029, while US 8,637,575 has a maximum term of April 2, 2028 (each not including any available PTE). The European patent was validated in France, Germany, Italy, Netherlands, Spain, Switzerland, and the United Kingdom.
- Patent Family 3 was filed on December 27, 2011 (PCT filing date). The priority date is December 27, 2010. This family is directed to compounds having a benzo[e][1,3]thiazin-7-one core, and methods of using these compounds as chemotherapeutic agents for the treatment of protein kinase related disorders, in particular cancer. National phase counterparts exist in Europe (EP 2658847) and the United States (US 9,073,880), both of which are now granted. EP 2658847 has a maximum term of December 27, 2031, while US 9,073,880 has a maximum term of April 9, 2032 (each not including any available PTE). The European patent was validated in France, Germany, Italy, Netherlands, Spain, Switzerland, and the United Kingdom.
- Patent Family 4 was filed on July 13, 2014 (PCT filing date). The priority date is July 14, 2013. This family is directed to use of the compounds disclosed in Patent Families 1-3, for the treatment of neurodegenerative diseases, including Alzheimer’s disease. National phase applications were filed in Europe (EP 3021944), the United States (US 9,770,454) and Israel (IL 243566). The European and Israeli applications are pending, while the U.S. patent is granted. Any patent issuing from this patent family will have a maximum patent term of July 13, 2034, not including any available PTE. A divisional application was filed in the U.S. and is now pending.
- Patent Family 5 was filed on February 4, 2016 (PCT filing date). The earliest priority date is February 5, 2015. This family is directed to combinations of the compounds disclosed in Patent Families 1-3, acting as dual modulators of Insulin Receptor Substrate (IRS) and signal transducer and activator of transcription 3 (STAT3), with various targeted drug families (inhibitors of Epidermal Growth Factor Receptor (EGFR), mammalian target of rapamycin (mTOR); mitogen-activated protein kinase (MEK) or mutated B-Raf), as well as chemotherapeutic agents (Gemcitabine, 5-FU, Irinotecan and Oxaliplatin), and use of such combinations for the treatment of cancer. The combinations can be used to treat tumors that have developed resistance to these anti-cancer drugs, to prevent acquired resistance of a tumor to these drugs, or to prevent tumor recurrence following cease of treatment with these drugs. The invention further relates to the treatment of cancer using combination therapy comprising a dual modulator of IRS and STAT3, in combination with an immunotherapy agent, and can be used to sensitize a tumor to immunotherapy. National phase applications were filed in Australia (AU2016213972), Brazil, Canada (CA2975673), China (CN107250108), Europe (EP3253733), India, Israel, Japan, Korea (KR20170109589), and the United States. Application numbers provided for published applications only. All of these applications are now pending. Any patent issuing from these applications will have a maximum patent term of February 4, 2036, not including any available PTE.
- Patent Family 6 was filed on November 16, 2017 (PCT filing date). There is no earlier priority date. This family is directed to specific combinations of the compounds disclosed in Patent Families 1 through 3 above, acting as dual modulators of certain anti-cancer mechanisms. The PCT application is currently pending, with an expected publication date of May 16, 2019. Any patent issuing from these applications will have a maximum patent term of November 16, 2037, not including any available PTE.
Exclusive License Agreement with Yissum
In August 2013, TyrNovo entered into a license agreement with Yissum Research and Development Company of the Hebrew University of Jerusalem Ltd. (“Yissum”), which was subsequently amended in April 2014 and March 2017, pursuant to which Yissum has granted TyrNovo an exclusive, license (with the right to sublicense) for the development, use, manufacturing and commercialization of products using certain patents and know-how owned by Yissum and patent applications filed by Yissum in connection with unique inhibitors of the IGF-1R Pathway (the “Yissum License Agreement”).
Under the terms of the Yissum License Agreement, Yissum shall retain the ownership of the Licensed Technology (as such term is defined therein). All rights in the results of the activities carried out by TyrNovo or third parties in the development of these products (and certain results obtained under material transfer agreements signed by TyrNovo and Yissum (the “TyrNovo MTAs”)) shall be solely owned by TyrNovo (unless an employee of the Hebrew University of Jerusalem or each of its branches is an inventor of any of the patents claiming such results, in which case they shall be owned jointly by Yissum and TyrNovo). TyrNovo has the right to grant sub-licenses to third parties in accordance with the terms set forth in the Yissum License Agreement.
TyrNovo has agreed to compensate Yissum for past patent expenses in a certain amount by no later than December 31, 2018. Yissum controls the prosecution, maintenance and enforcement of all the licensed patent rights. TyrNovo has the first right but not the obligation to take action against an infringement of a licensed patent right, if TyrNovo does not do so, Yissum may undertake such action at its own expense.
TyrNovo has agreed to pay Yissum a percentage of “net sales” as royalties and to pay Yissum a percentage of the income that it receives from granting sub-licenses to third parties. Additionally, in the event of an M&A prior to an IPO, TyrNovo will be required to pay Yissum a percentage of the proceeds received under such M&A. In the event of an IPO, then prior to the closing of such IPO. TyrNovo shall issue to Yissum such number of ordinary shares equal to a certain percentage of all TyrNovo shares.
Concurrent with the release of any of its ordinary shares that were issued to Katzenell Dimant Trustees Ltd. as trustee holding such shares in escrow on behalf of it and Goldman Hirsh Partners Ltd. (GHP) in connection with its acquisition during January 2017 of GHP’s controlling interest in TyrNovo, 12% of such ordinary shares are to be transferred by GHP to Yissum as payment for the share consideration portion of the exit fee by GHP under the Yissum License Agreement. 7,904,755 of its ordinary shares issued to the trustee are expected to be released in the coming days by the trustee to GHP, and 12% or 948,570 of such ordinary shares are to be concurrently transferred by GHP to Yissum as payment for the share consideration portion of the exit fee by GHP under the Yissum License Agreement. 3,387,753 Consideration Shares will then remain held in escrow in order to ensure the fulfillment of certain post-closing undertakings and to satisfy certain unresolved indemnification claims and other liabilities that the company may become subject to as a result of its acquisition of TyrNovo.
TyrNovo is required to indemnify Yissum, the Hebrew University of Jerusalem, their directors, employees, their executive officers, consultants or representatives and any other persons acting on their behalf under the license against any liability, including product liability, damages, losses, expenses, fees and reasonable legal expenses arising out of the TyrNovo’s actions or omissions or which derive from its use, development, manufacture, marketing, sale or sublicensing of any licensed product, licensed technology, and certain information obtained under the TyrNovo MTAs, or exercise of the Yissum License Agreement, and the TyrNovo MTAs.
TyrNovo has agreed to maintain, and to add Yissum as an additional insured party with respect to, clinical trials, comprehensive general liability and product liability insurance as well as an insurance policy with respect to the foregoing indemnification prior to the time when it commences clinical trials and concludes its first commercial sale.
The term of the Yissum License Agreement shall expire upon the later of ![]() the date of expiration in such country of the last to expire licensed patent included in the licensed technology; or (ii) the end of a period of 15 year of the first commercial sale in such country, while the license granted under the Yissum License Agreement will terminate upon the later of (unless the license has been earlier terminated or expired)
the date of expiration in such country of the last to expire licensed patent included in the licensed technology; or (ii) the end of a period of 15 year of the first commercial sale in such country, while the license granted under the Yissum License Agreement will terminate upon the later of (unless the license has been earlier terminated or expired) ![]() the date of expiration in such country of the last to expire licensed patent included in the licensed technology; (ii) the date of expiration of any exclusivity on the product granted by a regulatory or government body in such country; or (iii) the end of a period of 15 year of the first commercial sale in such country.
the date of expiration in such country of the last to expire licensed patent included in the licensed technology; (ii) the date of expiration of any exclusivity on the product granted by a regulatory or government body in such country; or (iii) the end of a period of 15 year of the first commercial sale in such country.
TyrNovo has the right to terminate the Yissum License Agreement upon a prior written notice. Either party has the right to terminate the Yissum License Agreement if the other party is in material breach and has not cured such material breach within a certain amount of days as of the receipt of a written notice notifying it of such breach. Additionally, Yissum has the right to terminate the Yissum License Agreement immediately in the event that TyrNovo does not comply with its obligation (following a certain amount of months cure period) to use commercially reasonable efforts to develop and commercialize the products; if an attachment is made over the majority of TyrNovo’s assets or if execution proceedings are taken against TyrNovo and are not set aside within a certain amount of days; or if TyrNovo challenges in any forum the validity of one or more of the licensed patents. Upon termination of the Yissum License Agreement, TyrNovo shall assign to Yissum all the results obtained during the development of the product. If Yissum licenses to third parties such results, then TyrNovo shall be entitled to a percentage of the net proceeds actually received by Yissum from such third parties, up to an amount covering TyrNovo’s expenses incurred during the development of such assigned results.
Market exclusivity
In the branded pharmaceutical industry, the majority of a branded drug’s commercial value is usually realized during the period in which the product has market exclusivity. In the U.S. and some other countries, when market exclusivity expires and generic versions of a product are approved and marketed, there can often be very substantial and rapid declines in the branded product’s sales. The rate of this decline varies by country and by therapeutic category, and the number of generic competitor entrants to the market, among other factors; however, following patent expiration, branded products often continue to have market viability based upon the goodwill of the product name, which typically benefits from trademark protection.
A pharmaceutical brand product’s market exclusivity is generally determined by two forms of intellectual property: patent rights held by the brand company and any regulatory forms of exclusivity to which the NDA-holder is entitled.
Patents are a key determinant of market exclusivity for most branded pharmaceuticals. Patents provide the brand company with the right to exclude others from practicing an invention related to the medicine. Patents may cover, among other things, the active ingredient(s), various uses of a drug product, pharmaceutical formulations, drug delivery mechanisms and processes for (or intermediates useful in) the manufacture of products, and polymorphs. Protection for individual products extends for varying periods in accordance with the expiration dates of patents in the various countries. The protection afforded, which may also vary from country to country, depends upon the type of patent, its scope of coverage and the availability of meaningful legal remedies in the country.
Market exclusivity is also sometimes influenced by regulatory exclusivity rights. Many developed countries provide certain non-patent incentives for the development of medicines. For example, the U.S., the European Union and Japan each provide for a minimum period of time after the approval of a new drug during which the regulatory agency may not rely upon the data of the original party who developed the drug to approve a competitor’s generic copy. Regulatory exclusivity rights are also available in certain markets as incentives for research on new indications, on orphan drugs and on medicines useful in treating pediatric patients. Regulatory exclusivity rights are independent of any patent rights and can be particularly important when a drug lacks broad patent protection. Most regulatory forms of exclusivity, however, do not prevent a competitor from gaining regulatory approval prior to the expiration of regulatory data exclusivity on the basis of the competitor’s own safety and efficacy data on its drug, even when that drug is identical to that marketed by the innovator.
It is not possible to predict the length of market exclusivity for any of its branded products with certainty because of the complex interaction between patent and regulatory forms of exclusivity, and inherent uncertainties concerning patent litigation. There can be no assurance that a particular product will enjoy market exclusivity for the full period of time that the company currently estimate or that the exclusivity will be limited to the estimate.




