Proteon Therapeutics
Overview
Proteon Therapeutics (PRTO) is a late-stage biopharmaceutical company focused on the development of novel, first-in-class pharmaceuticals to address the needs of patients with renal and vascular disease. The company's product candidate, vonapanitase, is a recombinant human elastase that Proteon Therapeutics is developing to improve vascular access outcomes in patients with chronic kidney disease, or CKD, undergoing or planning for hemodialysis, a lifesaving treatment that cannot be conducted without a functioning vascular access. The company believe data from its completed Phase 2 and Phase 3 clinical trials of vonapanitase support that a one-time, local application of investigational vonapanitase during surgical creation of a radiocephalic fistula for hemodialysis may improve fistula use for hemodialysis and secondary patency (time to fistula abandonment), thereby improving patient outcomes and reducing the burden on patients and the healthcare system.1
Arteriovenous fistulas are the gold standard of vascular access for hemodialysis, given they are associated with fewer complications and reduced rates of hospitalization as compared to other forms of vascular access. The company estimate there are approximately 130,000 fistulas created in the United States annually, a procedure in which a surgeon transects a vein and sutures it to the side of a nearby artery, typically in the arm. Radiocephalic fistulas created in the forearm are the preferred form of arteriovenous fistula because they preserve the maximum number of future sites for vascular accesses (i.e., the potential use of other vascular access sites further up in the arm) and are associated with the lowest rate of serious complications such as steal syndrome (hand ischemia).
However, radiocephalic fistulas suffer from a high rate of failure, typically due to insufficient blood flow, precluding hemodialysis. Recent data demonstrate that within one year of surgical creation of a radiocephalic fistula:
- Up to 55% will fail to become usable for hemodialysis because the fistula either has inadequate blood flow or cannot be successfully cannulated;
- Up to 40% will be abandoned (secondary patency loss); and
- Up to 70% will experience either a thrombosis or undergo a corrective procedure to restore or maintain blood flow (primary patency loss).
The need to improve vascular access outcomes for patients is well established in the hemodialysis community. Achieving a usable fistula for vascular access and maintaining the patency (blood flow) of such fistula enables the hemodialysis patient to avoid use of a dialysis catheter, the worst form of vascular access because of the increased risk of serious infection, hospitalization and death. Patients with a usable fistula also avoid the need for additional surgical procedures to create new forms of vascular access. Finally, as each patient has only a limited number of vascular access sites, fistula abandonment increases the risk that the patient may exhaust all permanent access sites and be subjected to chronic use of a dialysis catheter.
Vascular access failure also results in substantially higher healthcare costs. A recent study indicated that the total cost to Medicare for managing hemodialysis vascular access was more than $2.8 billion in 2013. This amount excludes costs of managing vascular access in predialysis patients and dialysis patients covered by Medicare Advantage HMOs or other non-Medicare payers, as well as patient co-pays and deductibles. It was also reported that fistulas that fail to become usable resulted in incremental costs to Medicare of up to $24,000 in the first year and $11,000 in the second year following fistula creation.
Because the clinical implications of fistula non-use and abandonment are severe, health care providers are aggressive in monitoring and intervening upon fistulas in an attempt to increase fistula use and reduce the rate of fistula abandonment. In less than a decade, the rate of procedures has approximately doubled. The function of usable fistulas can usually be restored via corrective procedures, either an intervention such as angioplasty, which is dilation of a blood vessel with a balloon, or a surgical revision. These procedures, however, are invasive, painful and associated with a number of complications. These procedures are also costly and often need to be repeated. Fistula patients in the United States on average require greater than 1.5 procedures per year, each of which typically costs Medicare between $5,000 and $15,000.
Proteon Therapeutics has demonstrated in nonclinical studies that vonapanitase fragments elastin fibers in blood vessels. Elastin fragmentation generates peptides in the adventitia that are recognized by cells possessing elastin receptors, including cells involved in vascular remodeling and the formation of neointimal hyperplasia. The generation of peptides in the adventitia may stimulate cells that restructure the vessel wall, promoting outward vascular remodeling, a process necessary for a fistula to become usable. In experimental models, fragmentation of elastin has been shown to be an early and essential event in outward vascular remodeling. The peptides are also chemo-attractants, which may reduce cell migration to the vessel lumen, inhibiting neointimal hyperplasia, which is the growth of tissue inside vessels that narrows fistulas and reduces blood flow. Vonapanitase has also been shown to lead to vessel dilation when administered at a sufficient concentration. During fistula creation surgery, a surgeon would administer drops of vonapanitase onto the surface of the artery and vein of a fistula for 10 minutes followed by a saline irrigation. Based on clinical and nonclinical studies, the company believe that a one-time, local application of investigational vonapanitase to the external surface of the vessels during fistula surgical creation may enhance outward vascular remodeling and inhibit neointimal hyperplasia, thereby reducing the risk of fistula failure.
PATENCY-1, the first of two multicenter, randomized, double-blind, placebo-controlled Phase 3 clinical trials, evaluated the safety and efficacy of a single dose of investigational vonapanitase in patients with CKD who underwent surgical creation of a radiocephalic arteriovenous fistula for hemodialysis. The company reported top-line data from the PATENCY-1 trial in December 2016. While this trial did not meet the primary endpoint of improving primary unassisted patency, Proteon Therapeutics is encouraged that vonapanitase demonstrated clinically meaningful improvements in other efficacy endpoints, including:
- 45% relative increase (20% absolute increase) in the proportion of patients who had a fistula that was used for hemodialysis (p=0.006);
- 34% reduction in the risk of secondary patency loss (fistula abandonment) over 12 months (p=0.048); and
- 56% relative increase (14% absolute increase) in the proportion of patients who had a fistula that was used for hemodialysis without prior corrective procedures such as angioplasty (p=0.035).
Reported adverse events were also comparable between the vonapanitase and placebo arms of the study. These events were consistent with the medical events experienced by patients with CKD undergoing surgical creation of a radiocephalic fistula.
The company's ongoing Phase 3 clinical trial, PATENCY-2, is the second Phase 3 trial of investigational vonapanitase in patients with CKD undergoing surgical creation of a radiocephalic fistula for hemodialysis. After announcing top-line results from the PATENCY-1 trial in December 2016, the company had discussions with the U.S. Food and Drug Administration, or FDA, regarding changes to the PATENCY-2 trial. Following its review of the complete data sets from the PATENCY-1 trial and discussions with the FDA, the company amended the protocol for the PATENCY-2 trial. The protocol amendment reordered the endpoints for this ongoing trial, establishing fistula use for hemodialysis and secondary patency as co-primary endpoints. The protocol amendment also increased the planned enrollment for this trial from 300 to 500 patients, which the company subsequently increased to 600 patients. The increased sample size for the PATENCY-2 trial provides power to detect the differences observed in the PATENCY-1 trial for fistula use for hemodialysis and secondary patency of 98% and 88%, respectively, with a p-value ≤0.05 for each of the co-primary endpoints. In connection with these changes, the company received written confirmation from the FDA that, if PATENCY-2 is successful in showing statistical significance (p≤0.05) on each of the co-primary endpoints, the PATENCY-2 trial together with data from previously completed studies would provide the basis for a Biologics License Application, or BLA, submission as a single pivotal study, in which case no additional studies would need to be conducted prior to submitting the BLA. The company completed enrollment in the PATENCY-2 trial in March 2018 and expect to report top-line data in March 2019. If the PATENCY-2 trial is successful, the company expect to submit a BLA in 2019.
Vonapanitase received Breakthrough Therapy designation from the FDA in May 2017 for hemodialysis vascular access indications. The FDA awards Breakthrough Therapy designations to expedite the development and review of investigational drugs that are intended to treat serious or life-threatening conditions when preliminary clinical evidence indicates that the treatment may offer a substantial improvement over currently available therapies on one or more clinically significant endpoints. Vonapanitase previously received Fast Track designation from the FDA which is also designed to facilitate the development and expedite the review of drugs and biologics to treat serious conditions and address an unmet medical need. The company also received orphan drug designations for vonapanitase in the United States and European Union for hemodialysis vascular access indications.
The company believe that, if its ongoing Phase 3 clinical trial is successful and vonapanitase is approved, vonapanitase will potentially become the standard of care for patients with CKD undergoing surgical creation of a radiocephalic fistula. The company retain worldwide commercial rights to vonapanitase. If approved by regulatory authorities, the company intend to commercialize this product in the United States itself with a specialty sales force, focused primarily on vascular surgeons. The company also intend to seek one or more collaborators to commercialize the product in additional markets, including Europe and China. The company's patents include claims covering formulations, methods of manufacturing and use of elastases, providing protection in the United States into 2030 and European Union through 2028, with potential extension into 2033 in the United States and in the European Union.
Strengths
The company believe its company and vonapanitase possess the following attributes that increase the likelihood that the company will be successful in developing and commercializing vonapanitase:
- Completed enrollment in pivotal Phase 3 trial. The company completed enrollment in the PATENCY-2 trial in March 2018 and expect to report top-line data in March 2019. Previously, the company made important changes to the protocol for the PATENCY-2 trial based on the results of its first Phase 3 trial, PATENCY-1. The protocol amendment for the PATENCY-2 trial reordered the existing endpoints, establishing co-primary endpoints of fistula use for hemodialysis and secondary patency (time to fistula abandonment), each of which demonstrated improvements in the PATENCY-1 trial. The company also increased the planned enrollment for the PATENCY-2 trial from 300 to 600 patients. The increased sample size for the PATENCY-2 trial provides power to detect the differences observed in the PATENCY-1 trial for fistula use for hemodialysis and secondary patency of 98% and 88%, respectively, with a p-value ≤0.05 for each of the co-primary endpoints. Proteon Therapeutics has also received written confirmation from the FDA that, if PATENCY-2 is successful in showing statistical significance (p≤0.05) on each of the co-primary endpoints, the PATENCY-2 trial together with data from previously completed studies would provide the basis for a BLA submission. If the PATENCY-2 trial is successful, the company expect to submit a BLA in 2019.
- Safety profile supports approval. Based on results from its clinical trials and preclinical studies, the company believe investigational vonapanitase, which is administered once and only acts locally, has demonstrated a favorable safety profile. Vonapanitase is applied to the outside of blood vessels for ten minutes and then washed away, which limits the potential for any absorption into the body and systemic activity. Any vonapanitase that might enter the bloodstream would be inactivated by anti-proteases, substances in the blood that inhibit the activity of vonapanitase. In clinical trials, there were no material increases in adverse events in the vonapanitase treatment groups as compared to placebo and no material findings related to physical examinations or clinical laboratory testing including chemistry, hematology and antibodies to vonapanitase. At its end of Phase 2 meeting with the FDA in 2013, the company confirmed that the company do not need to conduct any additional preclinical studies to support a BLA submission.
- Expedited programs to address unmet medical need. Vonapanitase has received Breakthrough Therapy and Fast Track designations from the FDA, which are designed to expedite the development and review of drugs and biologics to treat serious or life-threatening conditions and fill an unmet medical need. While radiocephalic fistulas are considered the preferred form of vascular access by the medical community, they are associated with high failure rates, most critically fistula non-use and abandonment. Achieving a usable fistula and maintaining patency (blood flow) enables the patient to avoid the temporary or permanent use of a dialysis catheter, the worst form of vascular access because of the increased risk of serious infection, hospitalization and death. Patients with a usable fistula also avoid the adverse effects of under-dialysis and the need for additional surgical procedures to create new fistulas. Proteon Therapeutics is not aware of any products approved in the United States or Europe that would compete with vonapanitase for the improvement of fistula use for hemodialysis and secondary patency.
- Substantial and readily-addressable market opportunity. If vonapanitase is approved, the company intend to commercialize this product in the United States itself with a specialty sales force, focused primarily on vascular surgeons. The company also intend to seek one or more collaborators to commercialize the product in markets outside of the United States, including Europe and China. In the United Sates, the company estimate a sales force of approximately 75-100 representatives will enable it to call on the approximately 1,300 hospitals that account for more than 90% of the fistula surgical creations performed annually. The company believe vonapanitase will be supported by key stakeholders, including referring nephrologists, patient advocacy groups, large dialysis organizations and payors. The company also believe that, if its ongoing Phase 3 clinical trial is successful and vonapanitase is approved, it will potentially become the standard of care for patients with CKD undergoing surgical creation of a radiocephalic fistula by increasing the use of fistulas for hemodialysis and reducing the rate of fistula abandonment and, in doing so, reduce the overall burden on patients and the healthcare system. The company also believe vonapanitase will be reimbursed adequately by Medicare, Medicaid and other public and commercial payors. Costs related to fistula surgical creation, which is typically performed in the hospital outpatient setting, are not included in the End Stage Renal Disease, or ESRD bundle, the single bundled payment from Medicare for a number of the costs of hemodialysis treatments, medications, labs and supplies for patients with end-stage renal disease. Vascular access failure results in substantially higher healthcare costs. A recent study indicated that the total cost to Medicare for managing hemodialysis vascular access was more than $2.8 billion in 2013, which excludes costs of managing vascular access in predialysis patients and dialysis patients covered by Medicare Advantage HMOs or other non-Medicare payers as well as patient co-pays and deductibles. It was also reported that fistulas that fail to become usable resulted in incremental costs to Medicare of up to $24,000 in the first year and $11,000 in the second year following fistula creation.
- Experienced team. The company's executive management team has extensive experience in the renal and vascular disease fields through their substantial involvement in companies such as Abbott, AMAG, GelTex, Genzyme, Glaxo and Merck. The company's Chief Executive Officer and Chief Medical Officer were senior executives at GelTex, a biopharmaceutical company, where they played leading roles in the development and commercialization of Renagel, a treatment for hemodialysis patients that led to Genzyme's acquisition of GelTex for more than $1 billion. The company's Senior Vice President of Commercial was a senior executive at AMAG Pharmaceuticals, a biopharmaceutical company, where he played a leading role in the commercialization of Feraheme for iron-deficiency anemia in adults with CKD.
Strategy
The company's strategy is to develop and commercialize vonapanitase for patients suffering from renal and vascular diseases, beginning with patients with CKD undergoing surgical creation of a radiocephalic fistula. Key elements of its strategy include its plans to:
- Complete clinical development of vonapanitase and seek regulatory approval in the United States in its lead indication. The company completed enrollment in the PATENCY-2 trial, its second Phase 3 trial for patients with CKD, in March 2018 and expect to report top-line data in March 2019. If PATENCY-2 is successful, the company expect to submit a BLA for vonapanitase to the FDA in 2019.
- Commercialize vonapanitase directly in the United States. If vonapanitase is approved by the FDA, the company intend to commercialize it itself in the United States with a specialty sales force focused primarily on vascular surgeons. Based on various third-party sources, the company estimate that approximately 130,000 arteriovenous fistulas are created annually. The company believe a specialty sales force of approximately 75-100 representatives will enable it to call on the approximately 1,300 hospitals that account for more than 90% of the fistula surgical creations performed in the United States annually. The company believe that, if its current Phase 3 clinical trial is successful and vonapanitase is approved, it will potentially become the standard of care for patients with CKD undergoing surgical creation of a radiocephalic fistula.
- Establish partnerships for the development and commercialization of vonapanitase in all or parts of Europe and other countries outside of the United States. Proteon Therapeutics is currently evaluating its existing clinical program to support filing in Europe. The company may, based on additional data including the data from its Phase 3 clinical trials in the United States and if sufficient funds become available, choose to undertake clinical development of vonapanitase in Europe. The company estimate that there are approximately 315,000 hemodialysis patients in Europe. The company plan to formally seek guidance from the European Medicines Agency, or EMA, in 2018 regarding its requirements for regulatory approval. If the company decide to conduct a clinical trial of vonapanitase in Europe or if such a clinical trial is necessary for regulatory approval, the company expect results from this trial to be available two to three years after the first patient is enrolled. Depending on the guidance obtained from the EMA and additional clinical data including data from the PATENCY-2 trial, the company would expect to submit a Marketing Authorization Application, or MAA. The company intend to seek one or more collaborators to develop and commercialize the product in European countries. In addition, the company may enter into collaborations for the development and commercialization of vonapanitase in other countries outside of the United States with large populations of hemodialysis patients, such as China and Japan. The company estimate that there are approximately 385,000 patients on hemodialysis in China, 320,000 patients on hemodialysis in Japan and more than 2,600,000 hemodialysis patients worldwide, with an annual worldwide growth rate of 6-7%. Approximately 90% of hemodialysis patients in China and Japan dialyze using an arteriovenous fistula.
- Pursue additional vascular access indications for vonapanitase. The company believe that its clinical data support further development of vonapanitase in brachiocephalic fistula creation. The company may, based on additional data including the data from its Phase 3 clinical trials and if sufficient funds become available, study the effects of a 30 microgram dose of vonapanitase versus placebo on brachiocephalic fistulas. If this trial were to successfully meet its primary endpoint, the company would expect to submit a supplemental BLA to the FDA and a supplemental MAA to the EMA. Further, if sufficient funds become available and after reviewing the results from its ongoing Phase 3 clinical trial, the company may commence a clinical trial of vonapanitase in patients undergoing placement of an arteriovenous graft. The company believe vonapanitase could offer a significant medical benefit in these patients.
- Pursue indications for vonapanitase in peripheral artery disease. In addition to vascular access indications, Proteon Therapeutics is investigating vonapanitase as a treatment for patients with symptomatic peripheral artery disease, or PAD. In 2016, the company initiated a Phase 1, multicenter, dose-escalation trial designed to evaluate the safety and technical feasibility of a single administration of vonapanitase as an adjunct to angioplasty for patients with PAD below the knee. The company expect to complete the enrollment and treatment of 24 patients in this study before the end of 2018. The company may, if sufficient funds become available, increase enrollment in the Phase 1 trial evaluating vonapanitase below the knee and/or begin patient enrollment in a Phase 1, multicenter, dose-escalation trial evaluating vonapanitase as a monotherapy for PAD above the knee.
- In-license or acquire additional product opportunities. The company plan to search for additional product opportunities that could be marketed and sold by the specialty sales force required to successfully launch vonapanitase in the United States if it is approved for marketing by the FDA.
Background on Hemodialysis
Healthy kidneys serve many functions, including eliminating metabolic waste products and excess water, helping to control blood pressure, and keeping electrolytes such as sodium and potassium in balance. Patients with CKD have lost kidney function, most commonly due to diabetes or hypertension. Kidney disease is progressive and once a patient has reached end-stage CKD, the kidneys are no longer able to perform their normal functions. At this point, some form of renal replacement therapy is required, such as hemodialysis, in which blood is processed by a hemodialysis machine; peritoneal dialysis, a process using a cavity in the abdomen called the peritoneum as a membrane across which fluids are exchanged from the blood; or kidney transplant.
Hemodialysis is the most common form of treatment for end-stage CKD. According to the U.S. Renal Data System 2017 Annual Data Report, in 2015 there were approximately 444,000 hemodialysis patients in the United States with approximately 109,000 new incident patients having started hemodialysis during the year, reflecting an annual U.S. growth rate of approximately 3%. The company believe that there are approximately 330,000 hemodialysis patients in Europe, 385,000 hemodialysis patients in China, 320,000 hemodialysis patients in Japan and 2.6 million hemodialysis patients worldwide, with an annual worldwide growth rate of 6-7%.
Hemodialysis is a chronic therapy performed by cannulating, or piercing, a vein with a large bore needle so that blood can be pumped through a hemodialysis machine, which removes metabolic waste and excess water normally excreted by the kidney. The cleansed blood is then returned to the same vein via a second needle. A hemodialysis session typically lasts three to four hours and is performed three times a week in an outpatient dialysis clinic.
To enable sufficient blood to pass through the hemodialysis machine to complete treatment within four hours, a vein must have blood flow of at least 500 milliliters per minute. The arm is the most convenient location for accessing the blood stream on a recurring basis, but blood flow in the arm is approximately 50 milliliters per minute. Therefore, most hemodialysis patients undergo a surgical procedure in which a surgeon establishes a direct connection between an artery and a vein, referred to as a fistula, to create a high flow circuit of sufficient diameter, most often in an arm. The fistula bypasses the capillary circulation in the hand and leads to a process known as maturation, where the internal diameter, or lumen, of the vein and blood flow increase over a period of weeks, resulting in a lumen diameter greater than 4 millimeters and blood flow of 500-2,000 milliliters per minute in successful cases.
Arteriovenous fistulas are the gold standard for vascular access. Arteriovenous fistulas are preferred because they are associated with fewer complications and reduced rates of hospitalization as compared to other forms of vascular access. As compared to grafts, fistulas require approximately 40% fewer interventional or surgical procedures and suffer from a rate of vascular access infection that is 54% lower. Patients dialyzing with a fistula have lower rates of thrombosis and hospitalization, longer survival, reduced mortality and lower cost of care. Beyond the substantial medical advantages of a fistula, available data from the U.S. Renal Data System show that patients who dialyze with a fistula cost Medicare approximately $15,000 less annually than patients who dialyze with a graft and approximately $25,000 less annually than patients who dialyze with a catheter. According to published data, approximately 68% of hemodialysis patients in the United States dialyze with a fistula compared to 67-83% of patients in the major European countries and approximately 90% of patients in China and Japan.
Based on various third-party sources, the company estimate there were approximately 130,000 fistulas created in the United States annually. There are a limited number of potential artery-vein combinations in the arm that can be used to create a fistula, principally the following:
- radiocephalic fistulas at the forearm (radial artery connected to cephalic vein), which the company estimate are created in 35 - 40% of new fistula creations;
- brachiocephalic fistulas at the elbow (brachial artery sutured to cephalic vein), which the company estimate are created in 50 - 55% of new fistula creations; and
- brachiobasilic fistulas in the upper arm (brachial artery sutured to basilic vein), which the company estimate are created in 10% of new fistula creations.
The medical community endorses radiocephalic fistulas as the optimal form of vascular access and the recommended first choice for new hemodialysis patients. Creating the vascular access site at the forearm preserves the potential future use of other accesses further up in the arm, is simpler to create, and is less likely to create heart failure due to very high blood flow or steal syndrome, where the diversion of flow through the fistulas reduces blood to the hand. Radiocephalic fistulas are also less likely to suffer from symptomatic central stenoses in the shoulder and chest, remote from the site of the fistula. The Kidney Disease Outcome Quality Initiative Guidelines, or KDOQI Guidelines, authored by the National Kidney Foundation, or NKF, specifically recommend starting with a radiocephalic fistulas if possible, stating that “starting [closer to the hand] and moving [further up the arm] provides for the possibility of preserving as many potential sites as possible for future access creation.” If a radiocephalic fistula must be abandoned, a surgeon can create a new vascular access higher up the arm, most likely a brachiocephalic fistula. However, if a brachiocephalic fistula is created first, the surgeon cannot later move down that same arm to create a radiocephalic fistula because the cephalic vein has already been transected for use in the brachiocephalic fistula.
While radiocephalic fistulas are considered the most desirable form of vascular access, radiocephalic fistulas suffer from high rates of patency loss and non-use for hemodialysis. Up to 40% of radiocephalic fistulas are abandoned within 12 months after their surgical creation. Additionally, up to 55% of radiocephalic fistulas fail to become usable for hemodialysis. Some patients never receive a radiocephalic fistula because the surgeon believes the risk of failure is too high for those patients. These patients who tend to be older and sicker will undergo creation of a fistula higher up on the arm and permanently lose at least one of their access sites. The company believe that the number of radiocephalic fistulas created annually may rise if vonapanitase improves outcomes and allows vascular surgeons to create radiocephalic fistulas in patients that they previously considered to be at an unacceptably high risk of failure.
The second choice for vascular access after a fistula is an arteriovenous graft in which a surgeon connects an artery and vein using a synthetic tube. Approximately 20% of hemodialysis patients in the United States dialyze with a graft, compared to approximately 5-12% of patients in the major European countries and approximately 2% and 7% of patients in China and Japan, respectively.
The least desirable type of vascular access is a catheter, a plastic tube that is placed directly through the skin into a vein, typically via an incision in the neck enabling placement of the catheter into a large vein that leads directly to the heart. The catheter connects the patient's vasculature to the hemodialysis machine. Because the catheter penetrates the skin continuously, it is subject to a high risk of infection and increased mortality. One of the primary goals of hemodialysis care is to keep patients off catheters. However, in the United States approximately 80% of patients initiate hemodialysis through a catheter until a fistula or graft is ready to be used, and are dialyzed through a catheter when the fistula or graft does not become usable for hemodialysis or must be abandoned and a new one has to be created. Approximately 12% of hemodialysis patients in the United States dialyze with a permanent catheter, compared to 10-28% of patients in the major European countries and approximately 10% and 2% of patients in China and Japan, based on published data.
Established Medical Need
The need to improve vascular access outcomes is well established in the hemodialysis community. The health-related and economic costs of creating vascular accesses and addressing access dysfunction and associated complications have led to a global effort to address the problem. Over the last twenty years, the NKF has established guidelines in an effort to increase the use of fistulas while reducing the rate of complications, mostly through the identification and promulgation of best practices. The National Institutes of Health, or NIH, joined the effort in 2000 with the creation of a multi-center consortium of medical centers, the Dialysis Access Clinical Trials Consortium, to coordinate the testing of new treatments designed to improve fistula and graft outcomes. The intensity of these efforts increased markedly in 2004, when the Centers for Medicare and Medicaid Services, or CMS, reacting to health and economic data, announced the “Fistula First” initiative to increase the use of fistulas while reducing complications. According to Fistula First, fistulas should be considered for every patient needing hemodialysis because, compared to other forms of vascular access, fistulas last longer, require fewer surgical and endovascular interventions, are associated with lower rates of infection, hospitalization and death, and are less costly. As a result of these efforts, fistula use has approximately doubled since 2004 to 68% of United States hemodialysis patients.
Although arteriovenous fistulas are the preferred form of vascular access, they suffer from a high rate of failure, typically due to insufficient blood flow, precluding hemodialysis. The increased use of fistulas has also led to a concurrent increase in the number of fistulas created in patients with higher risks of dysfunction. Manifestations of fistula failure can include the following:
- failure of the fistula to become usable for hemodialysis, in which the fistula either has inadequate blood flow or cannot be successfully cannulated;
- loss of primary patency, in which the fistula experiences a thrombosis or requires a corrective procedure to restore blood flow; or
- loss of secondary patency, in which the fistula is abandoned.
Proteon Therapeutics is not aware of products approved in the United States or Europe that would compete with vonapanitase for the improvement of fistula use for hemodialysis and secondary patency.
For patients on hemodialysis, fistula non-use or abandonment is associated with a number of poor outcomes, many of which are associated with prolonged catheter exposure. Patients whose fistula fails to become usable or is abandoned are often subjected to the following:
- Interrupted and missed dialysis sessions, which are associated with an increased risk of morbidity and mortality.
- Surgical placement of a new permanent access, often in the same arm but in a more proximal location, which is associated with increased complications such as steal syndrome (hand ischemia). A recent study indicated that subsequent accesses are at a greater risk of infectious and noninfectious complications compared to an initial access.
- New vascular accesses may also require additional corrective procedures to promote use of the fistula or maintain its function.
- Some patients may be considered poor candidates for a new fistula, resulting in placement of an arteriovenous graft, which are associated with higher rate of infection, thrombosis and patency loss compared to a fistula.
- Until a new permanent access is available for use, a process that typically requires a minimum of three months for fistulas in patients on hemodialysis, patients must dialyze with a catheter. If the new access fails to become usable, catheter exposure will be further lengthened. A recent study indicated that patients whose fistula fails to become usable are subjected to more than twice the time of catheter exposure in the first year after surgical creation of the fistula.
- Finally, some patients may be forced to dialyze with a catheter chronically, either because the patient has depleted all of his or her vascular access options or the patient refuses to undergo an additional surgical procedure to create a permanent access.
One of the primary goals of the Fistula First campaign is to reduce the use of catheters, which is considered the worst form of vascular access. As a foreign body, a catheter may incite chronic inflammation resulting in malnutrition, anemia and cardiovascular disease. Catheters are also subject to low blood flow, predisposing patients to underdialysis. In addition, patients dialyzing with a catheter are at heightened risk of hospitalization compared to patients dialyzing with a fistula. Patients dialyzing with a catheter average 18 hospital days per patient-year, which is approximately twice the rate of patients dialyzing with a fistula. Catheter exposure results in a substantially higher rate of infections, especially catheter-related bacteremia, due to the challenges of inserting and maintaining a foreign body through the skin. Infection is the second leading cause of death in hemodialysis patients, and catheter use is independently associated with increased risk of infectious, cardiovascular, and all-cause mortality compared to fistula use.
Among pre-dialysis patients, fistula failure can result in the patient initiating hemodialysis using a catheter instead of a fistula, which has been shown to be associated with increased risk of subsequent fistula failure and hospitalization. Patients who initiate hemodialysis on a catheter have two times the rate of admissions for infection in the first month of hemodialysis and a higher risk of all-cause hospitalization compared to patients initiating with a fistula. A recent study indicated an incremental cost increase of $30,000 in the first year of hemodialysis for patients initiating hemodialysis on a catheter.
Because the clinical implications of fistula non-use and abandonment are severe, health care providers are aggressive in monitoring and intervening upon fistulas in an attempt to increase fistula use and reduce the rate of fistula abandonment. In less than a decade, the rate of procedures has approximately doubled. Such procedures include balloon angioplasty and surgical revision, which are invasive, painful and associated with a number of complications. The procedures also often fail to provide a durable benefit, resulting in a cycle of interventions for the patient. Recent data indicate that 50% of fistulas that undergo angioplasty to treat patency loss experience another episode of patency loss within 12 months, resulting in the need for additional procedures to restore patency. Additionally, the procedures are not always successful in restoring patency, with up to 27% of the procedures failing to restore function, resulting in fistula abandonment. Patients in the United States using a fistula on average require more than 1.5 procedures per year, each of which typically costs Medicare between $5,000 and $15,000. A United States hospital published data in 2014 indicating that maintaining function in a radiocephalic fistula can cost on average more than $17,000 in the first year after surgical creation and in excess of $40,000 for the first and second year after surgical creation. In addition, a recent study indicated that the total cost to Medicare for managing hemodialysis vascular access was more than $2.8 billion in 2013. This amount excludes costs of managing vascular access in predialysis patients and dialysis patients covered by Medicare Advantage HMOs or other non-Medicare payers, as well as patient co-pays and deductibles. It was also reported that fistulas that fail to become usable resulted in incremental costs to Medicare of up to $24,000 in the first year and $11,000 in the second year following fistula creation.
Vonapanitase
Vonapanitase is a recombinant human elastase under development as a treatment to improve vascular access outcomes in patients with CKD undergoing or planning for hemodialysis, a lifesaving treatment that cannot be conducted without a functioning vascular access. Proteon Therapeutics has completed four multicenter, randomized, double-blind, placebo-controlled studies evaluating vonapanitase compared to placebo in patients with CKD, including the first of two Phase 3 clinical trials, PATENCY-1. The company also completed patient enrollment in its second Phase 3 clinical trial, PATENCY-2, in March 2018 and expect to release top-line data in March 2019. Vonapanitase received Breakthrough Therapy designation from the FDA in May 2017 for hemodialysis vascular access. The FDA awards Breakthrough Therapy designations to expedite the development and review of investigational drugs that are intended to treat serious or life-threatening conditions when preliminary clinical evidence indicates that the treatment may offer a substantial improvement over currently available therapies on one or more clinically significant endpoints. Vonapanitase previously received Fast Track designation from the FDA which is also designed to facilitate the development and expedite the review of drugs and biologics to treat serious conditions and address an unmet medical need. The company also received orphan drug designations for vonapanitase in the United States and European Union for hemodialysis vascular access indications.
Mechanism of Action
Based on clinical and nonclinical studies, the company believe that a one-time, local application of investigational vonapanitase to the external surface of the blood vessels during fistula surgical creation may enhance outward vascular remodeling and inhibit neointimal hyperplasia, thereby reducing the risk of fistula failure.
The company believe there are two primary causes of fistula failure:
- Failure due to insufficient outward vascular remodeling. For a fistula to become usable for dialysis, the outflow vein must outwardly remodel, experiencing an increase in diameter and blood flow. This process, known as vascular remodeling, involves proliferation of cells in the vessel wall, secretion of proteases that degrade extracellular matrix, and synthesis of new extracellular matrix by these cells. Fistulas that fail to outwardly remodel do not experience a sufficient increase in blood flow to enable successful dialysis.
- Failure due to neointimal hyperplasia. Fistulas may also fail to become usable for dialysis or may experience patency loss due to neointimal hyperplasia, which is scarring of the interior wall of the outflow vein near the lumen, resulting in stenosis of the vein lumen and obstruction of blood flow in the fistula. Neointimal hyperplasia typically occurs in response to vessel injury, such as following fistula creation or due to hemodynamic stress caused by the rapid flow of blood from the artery into the outflow vein. Fistulas also often undergo additional corrective procedures to restore blood flow, which expose the fistula to further mechanical injury. The response of the vein to these injuries results in activation and recruitment of scar forming cells, which multiply and migrate from the outside to the inside wall of the blood vessel and produce a layer of tissue, creating a narrowing in the vein lumen and reducing fistula blood flow. Fistulas that experience neointimal hyperplasia may not have sufficient blood flow to enable successful dialysis.
The company demonstrated through nonclinical studies that vonapanitase, at the concentrations being studied in arteriovenous fistulas, causes partial fragmentation of elastin fibers, primarily in the adventitial layer of the vessel wall. Elastin fragmentation generates peptides in the adventitia that are recognized by cells possessing elastin receptors, including cells involved in vascular remodeling and the formation of neointimal hyperplasia. The generation of peptides in the adventitia may stimulate cells that restructure the vessel wall, promoting outward vascular remodeling, a process necessary for a fistula to become usable. In experimental models, fragmentation of elastin is an early and essential event in outward vascular remodeling. The peptides are also chemo-attractants, which may reduce cell migration to the vessel lumen, focusing the healing response to vessel injury to the adventitial layer and inhibiting neointimal hyperplasia. In animal models of vascular injury, the porcine homologue of vonapanitase applied to the adventitial surface of veins and arteries fragmented elastin and directed the migration of proliferating cells away from the vessel lumen, significantly reducing neointimal hyperplasia. Vonapanitase has also been shown to lead to vessel dilation when administered at a sufficient concentration.
Clinical Development of Vonapanitase
The company's First Phase 3 Clinical Trial, PATENCY-1
In December 2016, the company announced that the PATENCY-1 trial did not meet its primary endpoint of improved primary unassisted patency compared to placebo (p=0.254). Primary unassisted patency was defined as the length of time from fistula surgical creation to the first occurrence of a fistula thrombosis or corrective procedure to restore or maintain patency (blood flow). While not statistically significant, vonapanitase treated patients demonstrated a 17% reduction in the risk of primary unassisted patency loss over one year. At the end of one year, 42% of vonapanitase-treated patients maintained primary unassisted patency, compared to 31% of placebo-treated patients. Median patency, the time at which 50% of patients in a group lost primary unassisted patency, was 171 days in the placebo group and 214 days in the vonapanitase treatment group based on the Kaplan-Meier estimates.
Kaplan-Meyer curves for primary unassisted patency in PATENCY-1:
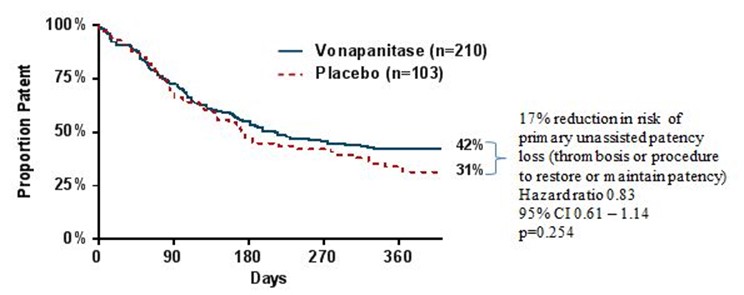
Secondary patency, the secondary endpoint in the PATENCY-1 trial, was defined as the length of time from surgical creation until fistula abandonment. Results suggested that vonapanitase may have improved secondary patency compared to placebo, as vonapanitase-treated patients demonstrated a 34% reduction in the risk of secondary patency loss (p=0.048). At the end of one year, 74% of vonapanitase-treated patients maintained secondary patency, compared to 61% of placebo-treated patients.
Kaplan-Meyer curves for secondary patency in PATENCY-1:
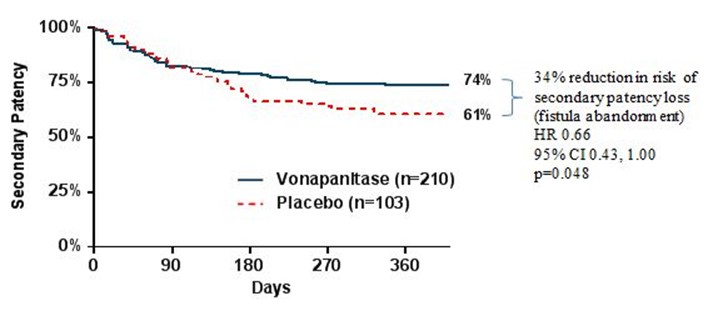
Results also suggested that vonapanitase may have improved fistula use for hemodialysis, one of the PATENCY-1 trial’s tertiary endpoints. 39% of vonapanitase-treated patients achieved unassisted use of their fistula for hemodialysis, compared to 25% of placebo-treated patients (p=0.035). 64% of vonapanitase-treated patients used their fistula for dialysis (unassisted or assisted), compared to 44% of placebo-treated patients (p=0.006). Use for hemodialysis was defined as use of the fistula for hemodialysis for at least 90 days or, if hemodialysis was not initiated at least 90 days prior to the patient’s last visit, for at least 30 days prior to the patient’s last visit and in use at the patient’s last visit. Unassisted use was defined as use without prior loss of primary unassisted patency.
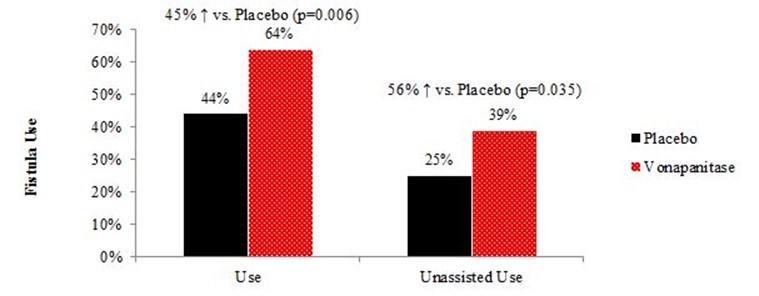
Results from the PATENCY-1 trial’s other tertiary endpoints include the following:
- Unassisted and Assisted Maturation. 63% of vonapanitase-treated patients achieved unassisted maturation, compared to 53% of placebo-treated patients (p=0.109). Unassisted maturation by ultrasound criteria was defined as achieving a vein diameter ≥4 millimeters and blood flow ≥500 milliliters per minute by three months without loss of primary patency. 66% of vonapanitase-treated patients achieved maturation without regard for prior procedures (i.e., unassisted or assisted) compared to 58% of placebo-treated patients (p=0.170).
- Rate of Procedures. Over one year, the rate per patient per year of procedures to restore or maintain patency was 1.10 in vonapanitase-treated patients compared to 1.48 in placebo-treated patients (p=0.479). Such procedures included thrombectomy, angioplasty, stent deployment and surgical revision. The rate per patient per year of all procedures was 1.54 in vonapanitase-treated patients compared to 2.07 in placebo-treated patients (p=0.149).
Proteon Therapeutics is continuing to follow patients who completed 12 months of follow-up in the initial trial with a patent fistula and consented to be enrolled in a patient registry to obtain long-term follow-up efficacy information.
Safety Data
Vonapanitase is administered topically at the vascular access and only acts locally. Proteon Therapeutics has not observed systemic activity or systemic toxicity in its preclinical animal studies, even following single-dose intravenous administration at very high multiples of the Phase 3 clinical trial doses. Safety evaluations in Phase 2 and Phase 3 clinical trials included ascertainment of adverse events, physical examinations, ultrasounds of the fistulas and nearby vessels, vital signs and laboratory studies. In the PATENCY-1 trial, no significant safety signals were identified, and no patients were considered positive for anti-drug antibodies. Most patients reported adverse events, the most common of which are summarized in the table set forth below, as compared to placebo. These events were generally consistent with the medical events experienced by CKD patients undergoing fistula creation surgery.
Proportions of Patients in PATENCY-1 Experiencing Most Common Adverse Events
| Adverse Events | Vonapanitase N=209 | Placebo N=102 |
|---|---|---|
| Vascular stenosis | 0.383 | 0.402 |
| Fistula thrombosis | 0.196 | 0.265 |
| Hypoaesthesia (numbness) | 0.053 | 0.049 |
| Procedural pain | 0.048 | 0.059 |
Note: Includes any adverse event that occurred in at least 5% of patients in either treatment group.
The percentages of serious adverse events, or SAEs, was also similar between the treatment groups in the PATENCY-1 trial (placebo 14.7%, vonapanitase 13.9%). Individual SAEs were never reported by more than one (1%) placebo patient or two (1%) vonapanitase patients with the exception of pneumonia reported by three (2.9%) placebo patients and myocardial infarction reported by two (2.0%) placebo patients. No SAEs were related to study drug with the exception of one (0.5%) arteriovenous fistula thrombosis in a vonapanitase patient. The percentages of severe SAEs were similar between treatment groups (placebo 4.9%, vonapanitase 4.8%) as were the percentages with life-threatening SAEs (placebo 4.9%, vonapanitase 3.3%). Life-threatening SAEs included acute myocardial infarctions, one cardiac arrest, and two pneumonias in the placebo group and single occurrences of coronary artery disease, cardiac arrest, pulseless electrical activity, death, injury, hypoxic-ischemic encephalopathy, and shock in the vonapanitase group. Four (3.9%) placebo patients and seven (3.3%) vonapanitase patients died during the study. All 11 deaths were considered unrelated to study drug.
The company's Ongoing Phase 3 Clinical Trial, PATENCY-2
The company's ongoing Phase 3 clinical trial, PATENCY-2, is the second of two randomized, double-blind Phase 3 trials, comparing a 30 microgram dose of investigational vonapanitase to placebo. As in PATENCY-1, PATENCY-2 enrolled patients with CKD undergoing surgical creation of a radiocephalic fistula for hemodialysis. Patients were randomized 2:1, vonapanitase to placebo, and are being followed for a period of 12 months. In March 2018, the company completed enrollment of a total of 603 treated patients at 39 centers in the U.S. and Canada. The company expect to report top-line data in March 2019.
Following its review of the complete data sets from the PATENCY-1 trial and discussions with the FDA, the company amended the protocol for the PATENCY-2 trial. The protocol amendment reordered the existing endpoints for the study, establishing fistula use for hemodialysis and secondary patency (time to fistula abandonment) as co-primary endpoints, each of which demonstrated improvements in the PATENCY-1 trial. Other efficacy endpoints in the amended protocol include unassisted fistula use for hemodialysis, primary unassisted patency, unassisted fistula maturation by ultrasound criteria, fistula maturation by ultrasound criteria, the rate of procedures performed to the fistula, and the rate of procedures to restore or maintain fistula patency. The company also increased the planned enrollment for this study from 300 to 600 patients. The increased sample size for the PATENCY-2 trial provides power to detect the differences observed in the PATENCY-1 trial for fistula use for hemodialysis and secondary patency of 98% and 88%, respectively, with a p-value ≤0.05 for each of the co-primary endpoints. The company received written confirmation from the FDA that, if PATENCY-2 is successful in showing statistical significance (p-value≤0.05) on each of the co-primary endpoints, the PATENCY-2 trial together with data from previously completed studies would provide the basis for a BLA submission as a single pivotal study, in which case no additional studies would need to be conducted prior to submitting the BLA.
The company's Phase 2 Fistula Clinical Trial
In 2012, the company completed a multicenter, randomized double-blind, placebo-controlled Phase 2 trial of vonapanitase in 151 patients undergoing surgical creation of a radiocephalic fistula (n=67) or brachiocephalic fistula (n=84). Patients were treated with vonapanitase at doses of 10 or 30 micrograms or placebo at the time of fistula creation and were followed for up to 12 months. The primary efficacy endpoint was primary unassisted patency, defined as the time from surgical creation of the fistula to occurrence of a thrombosis or a procedure, such as angioplasty, to restore or maintain patency. In the primary analysis for all fistulas, the risk of primary patency loss was not significantly reduced versus placebo for vonapanitase at doses of 10 micrograms (HR, 0.69; 95% CI, 0.39-1.22) or 30 micrograms (HR, 0.67; 95% CI, 0.38-1.19). Median patency, based on the Kaplan-Meier estimates, was 224 days in the placebo group and greater than 365 days in each of the vonapanitase treatment groups, indicating that vonapanitase prolonged primary unassisted patency. Ninety-two patients with a patent fistula who completed 12 months of follow-up in the initial trial were followed in a registry to obtain additional data related to the efficacy endpoints. In this follow-up, the vonapanitase 30 microgram benefit on primary unassisted patency persisted out over a median of three years.
An analysis of the primary endpoint data revealed an uneven distribution in patency loss events in patients with a brachiocephalic fistula due to central stenosis in the shoulder and chest, remote from the site of a fistula. Central stenoses commonly exist prior to fistula creation and are unmasked following creation of brachiocephalic fistulas, which have higher blood flow than radiocephalic fistulas. These stenoses are unrelated to treatment with vonapanitase. To correct for this uneven distribution, the company conducted a non-prespecified analysis of the primary endpoint that excluded patency loss events due to central stenoses. This analysis demonstrated a significant reduction in the risk of primary unassisted patency loss in the 30 microgram vonapanitase dose group (HR 0.52; 95% CI, 0.28-0.97; P=0.04) compared to placebo.
The benefit of vonapanitase in the Phase 2 trial on primary unassisted patency was most pronounced in the subset of patients undergoing creation of a radiocephalic fistula. The risk of primary patency loss was significantly reduced by vonapanitase at doses of 30 micrograms versus placebo (HR, 0.37; 95% CI, 0.15-.91; p=0.02). The subset analysis of this endpoint for radiocephalic fistula patients receiving the 30 microgram dose, which was not pre-specified, showed a significant increase in median primary unassisted patency of more than 365 days as compared to 125 days in the placebo group. The apparent benefit of the vonapanitase 30 microgram dose on primary unassisted patency persisted for these patients in the two-year registry period.
As with the primary efficacy analyses, the company performed a number of prespecified and exploratory analyses of the data on additional efficacy endpoints, including secondary patency loss and unassisted use for hemodialysis. The company observed no significant differences in the risk of secondary patency loss, which was defined as abandonment of the fistula, in the overall fistula population in the Phase 2 trial. However, a trend toward prolonged secondary patency was seen in patients receiving radiocephalic fistulas. In this non-prespecified subset analysis, treatment with vonapanitase at doses of 30 micrograms was associated with a reduction of 73% in the risk of secondary patency loss. However, this reduction in the risk of secondary patency loss was not statistically significant (HR, 0.27; 95% CI, 0.006-1.29; p=0.08). Additionally, in a recent publication of three-year follow-up data from the Phase 2 trial, a trend toward increased fistula use for hemodialysis was seen in the patients receiving radiocephalic fistulas when applying the definition of use for hemodialysis from the Phase 3 clinical trials of vonapanitase. While the differences observed in this prospective analysis were not statistically significant, 80% of patients receiving radiocephalic fistulas in the vonapanitase 30 microgram group used their fistula for hemodialysis compared with 56% in the placebo group (p=0.14).
In the Phase 2 trial, patients treated with vonapanitase reported adverse events comparable to placebo. These events were consistent with the medical events experienced by patients with CKD undergoing fistula creation surgery. Based on the results of this Phase 2 trial and its end of Phase 2 meeting with the FDA in April 2013, the company selected the 30 microgram dose for further study in the Phase 3 trials.
The company's Phase 1/2 Fistula Clinical Trial
The company submitted an investigational new drug application, or IND, for vonapanitase as a treatment for patients undergoing fistula creation on April 30, 2008. The company's initial clinical trial of vonapanitase was a Phase 1/2, randomized, double-blind, placebo-controlled, dose-escalation safety and exploratory efficacy trial in 66 patients undergoing creation of a radiocephalic or brachiocephalic fistula. Patients were treated with vonapanitase at nine dose levels ranging from 3.3 micrograms to 9 milligrams or placebo at the time of fistula creation and were followed for up to one year. This trial did not meet its primary endpoint, an endpoint the company did not pursue in its Phase 2 and Phase 3 trials. However, doses of vonapanitase at 3.3, 10 and 33 micrograms were associated with a trend toward prolonged primary unassisted patency (secondary endpoint p=0.66 in the All Treated population and p=0.15 in the All Treated Minus 3 population), fewer procedures to restore or maintain patency (collected as supportive data) and less hemodynamically significant lumen stenosis (collected as supportive data) compared with placebo treated patients or patients treated with higher vonapanitase doses. Higher doses showed results similar to placebo and no dose met the primary efficacy endpoint with statistical significance. No dose-related increases in adverse events were observed in the trial. Based on the results of this trial, the company selected 10 microgram and 30 microgram doses for further study in the Phase 2 trial.
Preclinical Development
Proteon Therapeutics has conducted an extensive preclinical program to evaluate the safety and tolerability of single doses of vonapanitase administered locally in animal models of fistula and arteriovenous graft creation, by percutaneous and endovascular injection in animal models of PAD as well as intravenously. Proteon Therapeutics has conducted preclinical studies in multiple species at doses up to 50 milligrams of vonapanitase, which is over 1,500 times higher than the dose the company used in its Phase 3 clinical trials. The company observed no systemic activity or systemic toxicity for vonapanitase in any of its preclinical studies. The company observed no toxicity in any of the doses that the company subsequently studied or plan to study in its Phase 3 clinical trial in humans. Only local toxicity was observed at surgical sites at high doses (10 and 50 milligrams, which is over 300-1500 times higher than the dose Proteon Therapeutics is studying in its Phase 3 clinical trial). These changes were reversible, with normal wound healing observed at 14 days except at the highest (50 milligrams) dose, in which there were some mild persistent changes in the jugular vein and subcutaneous tissue. Normal wound healing was observed in all the fistula studies in rabbits at doses up to 10 milligrams and in all the arteriovenous graft studies in dogs and pigs at doses up to 20 milligrams (the highest doses tested).
Other Programs, Indications and Trials
Other Fistula Trials
European clinical program
Proteon Therapeutics is currently evaluating its clinical program to support filing in Europe. The company may, based on additional data including the data from its Phase 3 clinical trials in the United States and if sufficient funds become available, choose to conduct a clinical trial of vonapanitase in Europe. Prior to initiating a European clinical trial, the company plan to formally seek guidance from the EMA regarding their requirements for regulatory approval.
Brachiocephalic Fistula
The company believe that its clinical data supports further development of vonapanitase in brachiocephalic fistula creation. The company may, based on additional data, including the data from its ongoing Phase 3 clinical trial, and if sufficient funds become available, study the effects of a 30 microgram dose of vonapanitase versus placebo on brachiocephalic fistulas. Prior to initiation of this trial, the company expect to seek guidance from the FDA regarding trial design.
Arteriovenous Grafts
An arteriovenous graft is a synthetic tube to connect a vein and an artery placed in a surgical procedure. In 2012, the company completed a Phase 1/2 randomized, double-blind, placebo-controlled, dose-escalation trial in 89 patients undergoing placement of an arteriovenous graft. Patients were treated with placebo or eight different doses of vonapanitase ranging from 10 micrograms to 9 milligrams at the time of graft placement and were followed for up to one year. Those patients who had not lost secondary patency were subsequently enrolled in a registry to obtain additional follow-up information on the arteriovenous graft.
The primary outcome measure was safety. Adverse events were consistent with the medical conditions experienced by patients with CKD undergoing graft surgery and showed no significant differences between groups. Some of the data showed indications of efficacy, especially in secondary patency, which is an approvable endpoint for hemodialysis access, for the groups treated with vonapanitase at doses of 10 micrograms and 30 micrograms.
After reviewing the results from its second Phase 3 clinical trial, and if sufficient funds become available, the company may commence a clinical trial of vonapanitase in patients undergoing placement of an arteriovenous graft.
Peripheral Artery Disease
In addition to vascular access indications, Proteon Therapeutics is investigating vonapanitase as a treatment for patients with symptomatic peripheral artery disease, or PAD. Patients with lower extremity PAD suffer from stenosis formation in the arteries providing blood to the legs. These patients typically present with exercise-induced leg pain, a condition known as intermittent claudication. Patients with claudication are unable to adequately maintain their activities of daily living because they quickly experience pain that can be resolved only through rest. Severe cases result in critical limb ischemia, or lack of oxygen, and the possibility of amputation. PAD is a global problem affecting a large number of people throughout the industrialized world. Approximately 8 million Americans suffer from PAD.
Patients with early stage PAD typically undergo lifestyle management such as smoking cessation, weight reduction and/or diabetes management, and treatment with oral medications. Approximately 800,000 patients in the United States who do not respond to lifestyle management and have worsening symptoms undergo an endovascular procedure, typically balloon angioplasty with or without stenting or vein bypass surgery. While these procedures work acutely to restore blood flow, they suffer from poor long-term durability, resulting in the need for repeat procedures.
The company believe that vonapanitase may improve the outcomes associated with angioplasty procedures, resulting in prolonged intervention-free patency while reducing the need for implantation of a permanent stent. The company submitted an IND for vonapanitase as a treatment for PAD patients on April 9, 2012. The company's initial PAD clinical trial was a Phase 1, open-label, dose-escalation safety/technical feasibility trial in 14 patients undergoing balloon angioplasty of the superficial femoral or popliteal arteries in the leg above the knee. Following successful angioplasty, patients were treated with vonapanitase via an FDA-cleared, drug-delivery catheter that allows vonapanitase to be administered locally in the outer layer of the vessel wall. Patients were followed for up to 12 months. The study met its stated objectives, as data indicated that catheter-based treatment with vonapanitase was generally well-tolerated and technically feasible. In the fourth quarter of 2016, the company initiated another Phase 1 study of vonapanitase delivered via a drug-delivery catheter in symptomatic PAD patients undergoing angioplasty of an artery below the knee. The company expect to complete the enrollment and treatment of 24 patients before the end of 2018 in this Phase 1 study and to follow each of these patients for period of up to seven months.
The company also believe that vonapanitase may be an alternative to traditional angioplasty. Vonapanitase may be delivered via a percutaneous approach, in which a physician inserts a needle through the skin to inject vonapanitase to the artery around the area of blockage. The company believe that vonapanitase may dilate the artery, resulting in increased lumen artery diameter, higher blood flow, and an improvement in clinical symptoms. In the fourth quarter of 2016 the company initiated a Phase 1 study of vonapanitase delivered as a monotherapy in patients with a clinical diagnosis of PAD due to an atherosclerotic lesion in an artery above the knee. Based on its current operating plan, Proteon Therapeutics has decided not to begin patient enrollment at this time.
The company believe that vonapanitase may improve the outcomes associated with vein bypass surgery, resulting in prolonged intervention-free patency. During vein bypass surgery, a surgeon places a vein, typically obtained from the patient’s leg, as an alternative conduit for blood to flow around the area of blockage restoring direct flow to the lower leg and foot. The company believe that vonapanitase, administered to the outside of the vein concurrently with the surgery, may improve the outcomes associated with vein bypass surgery, resulting in prolonged intervention-free patency.
Manufacturing and Supply
The company depend on third-party contract manufacturers for the production of vonapanitase. The company's API is produced at its contract manufacturer, Lonza LTD, or Lonza, which is required to comply with the FDA’s Current Good Manufacturing Practice, or cGMP, regulations. Vonapanitase finished product is produced at its contract fill/finisher providers, Jubilant HollisterStier and Patheon Manufacturing Services, LLC (formerly DSM Pharmaceuticals, Inc.), which is required to comply with cGMP regulations.
The company used API manufactured at Lonza to create finished drug product that was used in its Phase 3 fistula clinical trials. The company also plan to manufacture API at Lonza for its commercial launch and future fistula trials. The company also have a separate 5 milligram formulation that was used in its completed Phase 1 fistula study, Phase 2 fistula study, Phase 1 arteriovenous graft study, and Phase 1 PAD study. The company plan to use this 5 milligram formulation in its Phase 1 PAD studies.
The company modified its finished product at Jubilant HollisterStier for its Phase 3 trials and potential commercial launch in order to facilitate ease of administration and fill and finish at the 30 microgram doses. The modified finished product is reconstituted with sterile water to create a dosing solution containing 30 micrograms of vonapanitase. The company demonstrated that the modified finished product had the same elastase activity using synthetic and natural elastin substrates and the same elastin removal from blood vessels following ex vivo treatments as the previous finished product. The modified finished product formulation was similar to the previous finished product formulation in maintaining the health and viability of live cells in culture. These data suggest the modified finished product will have the same efficacy and safety in clinical trials as the previous finished product.
Release and stability testing for API and drug product are performed at PPD, Inc. The tests indicate stability of at least five years for its API and at least two years for its drug product.
In anticipation of a potential BLA submission, the company plan to manufacture a minimum of three batches of API and of drug product as part of process validation and to test these batches for stability with a goal of establishing a commercial shelf-life of at least two years for finished product and a longer expiry for API.
Sales and Marketing
The company's commercialization strategy is to develop vonapanitase into a leading therapy worldwide for the treatment of fistulas in patients with CKD undergoing or planning for hemodialysis and to improve vascular access outcomes in patients with other vascular diseases. If the PATENCY-2 trial is successful, the company expect to be able to submit a BLA in 2019.
Proteon Therapeutics has not yet established a commercial infrastructure, however, its Chief Executive Officer and other members of its executive management team have significant commercial experience in the industry, including commercial launch experience in the renal market. The company intend to recruit an in-house specialty sales force in the United States focused on promoting vonapanitase. The company plan to target its marketing and sales efforts at vascular surgeons who create fistulas. There are approximately 2,800 vascular surgeons in the United States. The company believe a sales force of approximately 75-100 representatives, supported by reimbursement specialists and a medical affairs team, will enable it to call on the approximately 1,300 hospitals that account for more than 90% of the fistula creations performed in the United States annually.
The company believe that vonapanitase will be reimbursed appropriately by Medicare, Medicaid and private payers. Costs related to fistula surgical creation, which is typically performed in the hospital outpatient setting, are not included in the ESRD bundle.
If vonapanitase is approved by the EMA, the company expect to commercialize vonapanitase in Europe with one or more commercial partners. The company also may enter into collaborations for the development and commercialization of vonapanitase in China, Japan and other countries outside of the United States.
Intellectual Property
The company strive to protect and enhance the proprietary technology, inventions and improvements that are commercially important to its business, including seeking, maintaining and defending patent rights. The company also rely on know-how that may be important to the development of its business. The company additionally expect to rely on regulatory protection afforded through data exclusivity, market exclusivity and patent term extensions where available.
The company's commercial success may depend in part on its ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to its business, as well as its ability to defend and enforce its patents and to operate without infringing the valid enforceable patents and proprietary rights of third parties.
The company's ability to prevent third parties from making, using, selling, offering to sell or importing competing products to ours, including a competitor to vonapanitase, depends on the scope of its patents. Proteon Therapeutics has several patents and patent applications relating to the vonapanitase formulation and its therapeutic uses, and the company possess substantial know-how relating to the development and commercialization of vonapanitase. The company cannot be sure that any of its pending patent applications or future patent filings will lead to the issuance of new patents, nor can the company be sure that any of its existing patents or any patents that may be granted to it in the future will be adequate to protect its market.
The company plan on pursuing in-licensing opportunities to develop, strengthen and maintain its proprietary position for its products. The company expect to use trademark protection for its products as they are marketed.
Patents
As of December 31, 2017, the company own 37 issued patents and 20 pending patent applications. The patents and applications primarily fall into two families, a first relating to the vonapanitase formulation and its manufacture and use, as well as other formulations of elastases (the “formulation family”), and the second relating to certain therapeutic uses of vonapanitase, and associated systems and kits that include a catheter and are suitable for a subset of those therapeutic uses (the “therapy family”). The formulation family includes two issued United States patents, two issued European patents, additional patents issued in Australia, China, Hong Kong, India, Israel, Japan, Mexico, New Zealand, Russia, South Korea and Taiwan, and patent applications pending in several major jurisdictions worldwide, including Japan, China, South Korea, Brazil, Mexico, Europe and the United States. The expected expiration date for any patents that have issued or may issue from the formulation family is December 4, 2028, exclusive of possible patent term extension available for one patent covering vonapanitase under the Hatch-Waxman Amendments or comparable provisions in other jurisdictions, except in the United States where its two formulation family patents were awarded patent term adjustments of 199 and 668 days, respectively, due to United States Patent and Trademark Office, or USPTO delays taking their expiration dates to June 20, 2029 and October 3, 2030, respectively. The therapy family includes eight issued United States patents, three issued European patents, one issued Canadian patent, one issued Hong Kong patent, and an application pending in the United States. The expected expiration date for any patents that have issued or may issue from the therapy family patents is September 24, 2020, except in the United States where several patents were awarded a patent term adjustment and the expected expiration date of two therapy family patents related to systems and kits including elastase and a catheter is June 30, 2021, exclusive of possible patent term extension.
Patent Term
The base term of a U.S. patent is 20 years from the filing date of the earliest-filed non-provisional patent application from which the patent claims priority. The term of a U.S. patent can be lengthened by patent term adjustment, which compensates the owner of the patent for administrative delays at the USPTO. In some cases, the term of a U.S. patent is shortened by terminal disclaimer that reduces its term to that of an earlier-expiring patent.
The term of a U.S. patent may be eligible for patent term extension under the Hatch-Waxman Amendment, to account for at least some of the time a product is under development and regulatory review after the patent is granted. With regard to a product for which FDA approval is the first permitted marketing of the active ingredient, the Hatch-Waxman Act allows for extension of protection of one U.S. patent that includes at least one claim covering the composition of matter of an FDA-approved product, an FDA-approved method of treatment using the product, and/or a method of manufacturing the FDA-approved product. The extended protection cannot exceed the shorter of five years beyond the non-extended expiration of the patent or 14 years from the date of the FDA approval of the product. Some foreign jurisdictions, including Europe, have analogous patent extension provisions, which allow for extension of the protection of a patent that covers a drug approved by the applicable foreign regulatory agency. In the future, if and when vonapanitase receives FDA approval, the company expect to apply for patent extension to extend the protection of one of its patents covering vonapanitase or its use.
Assignment of Rights and License Agreement
As successor to Proteon Therapeutics, LLC by merger, the company acquired all of the assets of the LLC, including all of the intellectual property rights in a patent family entitled “Local, Transcatheter Delivery of Proteases to Reopen Obstructed Biological Conduits” (the “JHU patent family”). This patent family was originally developed by its founder, Dr. F. Nicholas Franano, at The Johns Hopkins University, or Johns Hopkins, and includes United States patent Nos. 7,063,838; 7,153,505; 7,361,335; 7,632,494; 7,883,699; 8,524,226; 8,562,983; and 8,568,716. Johns Hopkins assigned all of the intellectual property rights to Dr. Franano who in turn assigned the rights to the LLC. Under the terms of the assignment of rights and license agreement with Johns Hopkins, Dr. Franano reimbursed certain costs of Johns Hopkins and agreed to pay the future costs and expenses of patent prosecution and maintenance, as well as any costs related to infringement. In addition, under the agreement, Dr. Franano granted to Johns Hopkins rights to practice under the intellectual property rights for non-profit purposes. The company's rights are further subject to any rights the United States Government may have in inventions that are the subject matter of the acquired patents under the Bayh Dole Act due to its sponsorship of research that led to certain of such inventions. The agreement does not specify a term and does not include any termination provisions. Dr. Franano agreed that upon commercialization of the assigned invention, he would remit to Johns Hopkins 2.5% of any revenues or fees received from certain net sales of any product covered by the JHU patent family. The company assumed, and are the successor to, all of Dr. Franano's payment and other obligations to Johns Hopkins. Seven U.S. patents in the JHU patent family, and their foreign counterparts, described above as the therapy family, relate to certain therapeutic uses of vonapanitase, and the associated systems and kits that include a catheter and are suitable for a subset of those therapeutic uses.




