Puma Biotechnology
Overview
Puma Biotechnology (PBYI) is a biopharmaceutical company with a focus on the development and commercialization of innovative products to enhance cancer care. The company in-license the global development and commercialization rights to three drug candidates – PB272 (neratinib, oral), PB272 (neratinib, intravenous) and PB357. Neratinib is a potent irreversible tyrosine kinase inhibitor, or TKI, that blocks signal transduction through the epidermal growth factor receptors, HER1, HER2 and HER4. Currently, Puma Biotechnology is primarily focused on the U.S. commercialization of NERLYNX (neratinib), its first U.S. Food and Drug Administration, or FDA, approved product, and on the further development of the oral version of neratinib for additional indications in the treatment of HER2-positive breast cancer. The company believe neratinib has clinical application in the treatment of several other cancers as well, including non-small cell lung cancer and other tumor types that over-express or have a mutation in HER2. Until recently, Puma Biotechnology has focused its efforts and resources primarily on obtaining regulatory approval for NERLYNX (neratinib) and on acquiring and developing its pharmaceutical technologies, raising capital and recruiting personnel.1
On July 17, 2017, the company received regulatory approval of its first product, NERLYNX (neratinib), formally known as PB 2727 (neratinib (oral)), for the extended adjuvant treatment of adult patients with early state HER2-overexpressed/amplified breast cancer following adjuvant trastuzumab-based therapy from the FDA. After receiving FDA approval, the company commenced commercialization of NERLYNX in the United States using a direct sales force.
Before the company can market neratinib in countries outside the United States, the company must receive regulatory approval from the appropriate government entities in those countries. The company filed a Marketing Authorization Application, or MAA, with the European Medicines Agency, or EMA, in July 2016. The company recently announced that the EMA’s Committee for Medicinal Products for Human Use, or CHMP, adopted a negative opinion and recommend refusal of its MAA for neratinib for the extended adjuvant treatment of early stage HER2-positive breast cancer. Puma Biotechnology has 15 days from the date of acknowledgement of receipt of the final opinion package to request a re-examination, and the company intend to submit the request within the prescribed timeline. The company recently entered into exclusive license agreements with Specialised Therapeutics Asia Pte Ltd., or STA, Medison Pharma Ltd., or Medison, and CANbridgepharma Limited, or CANbridge, to pursue regulatory approval and commercialize NERLYNX, if approved, in South East Asia, Israel and greater China, respectively. The company plan to continue to pursue commercialization of NERLYNX in other countries outside the United States, if approved, and will evaluate various commercialization options in those countries, including developing a direct sales force, contracting with third parties to provide sales and marketing capabilities, or some combination of these two options. The company expect that its expenses will continue to increase as the company continue commercialization efforts.
Breast cancer is the leading cause of cancer death among women worldwide. Studies show that approximately 20% to 25% of breast cancer tumors have an over-expression of the HER2 protein. Women with breast cancer that over-expresses HER2, referred to as HER2-positive breast cancer, are at greater risk for disease progression and death than women whose tumors do not over-express HER2. Therapeutic strategies, such as the use of trastuzumab (marketed as Herceptin), pertuzumab (marketed as Perjeta) and T-DM1 (marketed as Kadcyla), each produced by Genentech, and lapatinib (marketed as Tykerb) produced by Novartis, given either alone or in combination with chemotherapy, have been developed to improve the treatment of this type of breast cancer by binding to the HER2 protein. There are a number of trials ongoing that involve various combinations of these drugs (for example, Perjeta). Based on pre-clinical studies and clinical trials to date, the company believe that neratinib may offer an advantage over existing treatments by more potently inhibiting HER2 at a different site and using a different mechanism than these other drugs.
Separately, in February 2013, the company reached agreement with the FDA under a Special Protocol Assessment, or SPA for a planned Phase III clinical trial of PB272 in patients with HER2-positive metastatic breast cancer who have failed two or more prior treatments (third-line disease). The EMA has provided follow-on scientific advice, or SA, consistent with that of the FDA regarding its ability to use the trial to support regulatory approval in the European Union. The company refer to this trial as PUMA-NER-1301. The company initiated this trial in June 2013. The company expect to report the top line data from this trial in 2018.
Additionally, in December 2016, the company initiated a managed access program for neratinib. Managed access programs provide physicians and patients access to medicines when there are limited or no other therapeutic options available. The company's managed access program for neratinib enables participation from countries outside the United States, including European Union member states, where permitted by applicable rules, procedures and regulatory authorities. The program will provide access to neratinib for the treatment of early stage HER2-positive breast cancer (extended adjuvant setting), HER2-positive metastatic breast cancer and HER2-mutated solid tumors. In order for patients to qualify for its managed access program they must be unable to participate in any ongoing neratinib clinical trial. Patients in the managed access program will be given neratinib and will be instructed to take a prophylaxis during treatment to manage neratinib-related diarrhea, which the company expect will consist of high dose loperamide and budesonide. Puma Biotechnology has partnered with Caligor Opco LLC, which specializes in early access to medicines, to implement and oversee the managed access program for neratinib.
In addition to continuing to follow the patients from the ExteNET trial and continuing the PUMA-NER-1301 trial, Puma Biotechnology is actively conducting the following trials to evaluate the safety and efficacy of neratinib in various indications:
- a Phase II clinical trial of neratinib for the extended adjuvant treatment of patients with early stage HER2-overexpressed/amplified breast cancer who have received prior adjuvant trastuzumab (Herceptin)-based therapy in which patients are given antidiarrheal prophylaxis including loperamide alone or in combination with budesonide or other agents in order to prevent and reduce the neratinib-related diarrhea;
- a Phase II clinical trial of neratinib in combination with the chemotherapy drug capecitabine in patients with HER2-positive metastatic breast cancer that has metastasized to the brain;
- a Phase II clinical trial of neratinib in combination with the endocrine therapy fulvestrant in the treatment of patients with HER2-negative breast cancer that have a HER2 mutation;
- a Phase II clinical trial of neratinib monotherapy in the treatment of solid tumors that have an activating EGFR exon 18, HER2 of HER4 mutation;
- a Phase II clinical trial in the treatment of HER2-mutated non-small cell lung cancer; and
- a Phase I/II trial of neratinib plus Kadcyla in patients with metastatic HER2-positive breast cancer.
The company license the commercial rights to its current drug candidates from Pfizer, Inc., Pfizer or the Licensor, which had previously been responsible for the clinical trials regarding neratinib. Going forward, the company expect to augment its product pipeline by acquiring, through license or otherwise, additional drug candidates for research and development, and potential commercialization. In evaluating potential drug candidates, the company employ disciplined decision criteria that favor drug candidates that have undergone at least some clinical study. The company's decision to acquire a drug candidate will also depend on its evaluation of the scientific merits of the underlying technology, the costs of the transaction and other economic terms of any proposed license, the amount of capital that the company anticipate will be required to develop the drug candidate and the economic potential of the drug candidate if approved for commercialization. The company believe this strategy minimizes its clinical development risk and allows it to accelerate the development and potential commercialization of current and future drug candidates.
Strategy
The company's primary objective is to build neratinib into a significant oncology franchise as a single agent, and potentially in combination with other therapies. The following elements comprise the strategy to achieve this objective:
- Seek regulatory approval and commence commercialization of neratinib in regions outside the United States. Before the company can market neratinib outside the United States for any indication, including the FDA-approved indication associated with NERLYNX, the company must obtain regulatory approval in those countries. The company recently entered into exclusive license agreements with STA, Medison, and CANbridge pursuant to which each will develop and commercialize NERLYNX in South East Asia, Israel, and greater China, respectively. Pursuant to these agreements the company receive upfront and milestone payments and will receive royalties on sales once commercialized. In June 2016, the company submitted an MAA to the EMA for neratinib for the extended adjuvant treatment of patients with early-stage HER2-overexpressed/amplified breast cancer who have received prior adjuvant trastuzumab-based therapy. The MAA submission was based upon the results of the ExteNET trial, which reached its primary endpoint whereby neratinib demonstrated a statistically significant reduction of risk of invasive disease recurrence or death versus placebo. The CHMP recently recommended refusal of its MAA for neratinib for the extended adjuvant treatment of early stage HER2-positive breast cancer but allowed it to request a re-examination, which the company intend to do. Puma Biotechnology is continuing to also evaluate potential commercialization options for the extended adjuvant setting in additional countries outside the United States, including developing a direct sales force, contracting with third parties to provide sales and marketing capabilities, some combination of these two options or other strategic options.
- Continue to advance the development of neratinib for the treatment of other HER2-positive or HER2 mutated breast cancer indications. Puma Biotechnology is primarily focused on developing neratinib for the treatment of patients with HER2-positive breast cancer, HER2-negative breast cancer with a HER2 mutation or other solid tumors with an activating mutation in HER2, or patients with HER2-mutated non-small cell lung cancer. In addition to its completed ExteNET trial, Puma Biotechnology has several ongoing clinical trials focused on the treatment of patients with HER2-positive breast cancer. In June 2013, the company commenced a Phase III clinical trial of neratinib in patients with HER2-positive metastatic breast cancer who have failed two or more prior treatments (third-line disease). The company also have several ongoing Phase I and Phase II clinical trials evaluating the use of neratinib in combination with various other drugs, including Kadcyla, Xeloda, Paclitaxel and Torisel, to treat patients with HER2-positive metastatic breast cancer and HER2-positive metastatic breast cancer that has metastasized to the brain.
- Expand its product pipeline by pursuing additional applications of neratinib. The company believe there are additional applications for neratinib in the treatment of HER2-mutated non-small cell lung cancer, which the company also believe may be underserved by current treatment alternatives; in the treatment of patients with HER2-negative breast cancer who have a HER2 mutation; and in tumor types where HER2 is over-expressed or mutated. The company intend to further evaluate the safety and efficacy of neratinib for treating these cancers.
- Build a sustainable product pipeline by employing multiple therapeutic approaches and disciplined decision criteria based on clearly defined proof of principal goals. The company seek to build a sustainable product pipeline by employing multiple therapeutic approaches and by acquiring drug candidates belonging to known drug classes. In addition, the company employ disciplined decision criteria to assess drug candidates, favoring drug candidates that have undergone at least some clinical study. The company's decision to license a drug candidate will also depend on the scientific merits of the technology; the costs of the transaction and other economic terms of the proposed license; the amount of capital required to develop the technology; and the economic potential of the drug candidate, should it be commercialized. The company believe this strategy minimizes its clinical development risk and allows it to accelerate the development and potential commercialization of current and future drug candidates. The company intend to pursue regulatory approval for a majority of its drug candidates in multiple indications.
- Evaluate the commercialization strategies on a product-by-product basis in order to maximize the value of each. Puma Biotechnology is currently commercializing NERLYNX using a direct sales force in the United States and using out-licenses in certain countries outside of the United States. As the company move additional drug candidates through development toward regulatory approval, the company plan to evaluate several options for each drug candidate’s commercialization strategy. These options include building its own internal sales force; entering into a joint marketing partnership with another pharmaceutical or biotechnology company, whereby the company jointly sell and market the product; and out-licensing its product, whereby another pharmaceutical or biotechnology company sells and markets its product and pays it a royalty on sales. The company's decision may be different for each product that reaches commercialization and will be based on a number of factors including capital necessary to execute on each option, size of the market to be addressed and terms of potential offers from other pharmaceutical and biotechnology companies.
Breast Cancer Overview
Breast cancer is the leading cause of cancer death among women worldwide, with approximately 1 million new cases reported each year and more than 400,000 deaths per year. Approximately 20% to 25% of breast cancer tumors show over-expression of the HER2 protein. Women with breast cancer that over-expresses HER2 are at greater risk for disease progression and death than women whose tumors do not over-express HER2. Therapeutic strategies have been developed to block HER2 in order to improve the treatment of this type of breast cancer.
Trastuzumab, pertuzumab, lapatinib and T-DM1 are all drugs that bind to the HER2 protein and thereby cause the cells to cease reproducing. Today, these drugs are used as single agents, in combination with other drugs and in combination with chemotherapy to treat patients with HER2-positive breast cancer at various stages.
Currently, the only treatment approved by the FDA for the treatment of neoadjuvant (newly diagnosed) HER2-positive breast cancer is the combination of pertuzumab plus trastuzumab and taxane chemotherapy. The FDA-approved therapy for the adjuvant treatment of HER2-positive early stage breast cancer is the combination of trastuzumab and chemotherapy. In addition, the combination of pertuzumab plus trastuzumab and chemotherapy was recently approved as adjuvant treatment of patients with HER2-positive early breast cancer at high risk of recurrence based on the results of the APHINITY trial. Puma Biotechnology is also aware of the KAITLIN trial, which is comparing trastuzumab plus pertuzumab plus taxane following anthracyclines versus T-DM1 plus pertuzumab following anthracyclines as an adjuvant therapy.
Trastuzumab and pertuzumab given in combination with taxane chemotherapy is the current first-line standard of care for HER2-positive metastatic breast cancer. Lapatinib (Tykerb), given in combination with the chemotherapy drug capecitabine, is also FDA-approved for the treatment of patients who have failed prior treatments. In a Phase II clinical trial, lapatinib demonstrated a median progression free survival of 8 to 9 weeks and a response rate of 5 – 7%. In a Phase III clinical trial, patients with HER2-positive metastatic breast cancer who received the combination of lapatinib plus capecitabine demonstrated a median progression free survival of 27.1 weeks and a response rate of 23.7%. In the Phase III EMILIA trial, the combination of lapatinib plus capecitabine demonstrated a median progression free survival of 25.6 weeks and a response rate of 30.8%. T-DM1 is approved by the FDA for the treatment of patients with HER2-positive metastatic breast cancer who previously received first line trastuzumab-based therapy. Unfortunately, the disease eventually progresses for most patients with HER2-positive breast cancer while on these treatments. For these reasons, there is a need for alternatives to block HER2 signaling in patients who fail treatment with prior HER2 directed treatments. Neratinib is an orally active small molecule that inhibits HER2 at a different site and uses a different mechanism than trastuzumab. As a result, the company believe that neratinib may have utility in patients with HER2-positive metastatic breast cancer who have failed treatment with trastuzumab.
The company believe that there are approximately 36,000 patients in the United States and 34,000 patients in the European Union, or the EU, with newly diagnosed HER2-positive breast cancer. Based on its internal estimates, the company believe that the worldwide Herceptin adjuvant revenue was approximately $4.5 to $5.0 billion in 2015. The company also believe that there are between 5,000 and 6,000 patients in the United States with third-line or later HER2-positive metastatic breast cancer. The number of patients with third line or later HER2 positive metastatic breast cancer may decrease in future years as the introduction of new neoadjuvant, adjuvant and extended adjuvant treatments may reduce the number of patients with recurrence of HER2 positive breast cancer and therefore reduce the number of patients with HER2 positive metastatic breast cancer. In 2013, worldwide sales of Tykerb for this indication were approximately $325 million.
The company believe that approximately 2% of all newly diagnosed breast cancer patients have mutation in HER2 kinase (approximately 4,000 to 5,000 patients in the United States) and that approximately 4 – 5% of all metastatic breast cancer patients have mutation in HER2 kinase (approximately 8,000 to 10,000 patients in the United States). The company believe that this mutation occurs mostly in patients with hormone receptor-positive disease.
Product Development Pipeline
The following chart shows each of its current drug candidates and their clinical development stage.
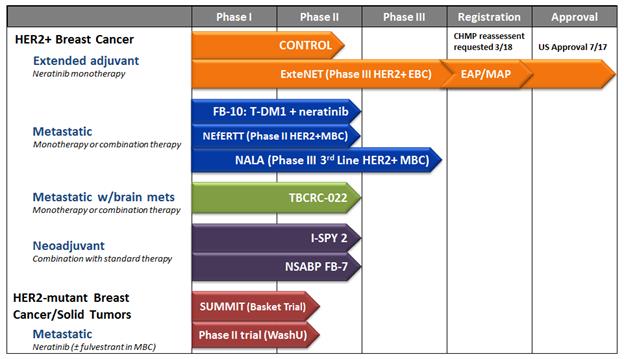
Neratinib
Neratinib is a potent irreversible tyrosine kinase inhibitor, or TKI, that blocks signal transduction through the epidermal growth factor receptors, HER1, HER2 and HER4. Based on pre-clinical studies and clinical trials to date, the company believe that neratinib may offer an advantage over existing treatments that are used in the treatment of patients with HER2-positive metastatic breast cancer who have failed prior treatments, including treatment with trastuzumab, pertuzumab, and T-DM1. Currently, the treatment of metastatic breast cancer patients involves treatment with these agents either alone or in combination with chemotherapy. The company believe that by more potently inhibiting HER2 at a different site and acting via a mechanism different from other agents, neratinib may have therapeutic benefits in patients who have failed these existing treatments, most notably due to its increased selectivity and irreversible inhibition of the HER2 target enzyme.
In addition, the company believe neratinib has clinical application in the treatment of other cancers, including non-small cell lung cancer and other tumor types that over-express or have a mutation in HER2.
The company's initial focus is on the development of the oral formulation of neratinib. Puma Biotechnology is also evaluating for potential development an intravenous formulation of neratinib and PB357, a back-up compound to neratinib.
PB272 (neratinib oral)—Early Stage Breast Cancer
tended Adjuvant Breast Cancer
Two-Year ExteNET Data. In July 2014, the company announced top line results from the Phase III clinical trial of neratinib for the extended adjuvant treatment of early stage HER2-positive breast cancer (ExteNET Trial). The data from this trial was presented in an oral presentation at the American Society of Clinical Oncology (ASCO) 2015 Annual Meeting in June 2015 and was published online in The Lancet Oncology in February 2016. The ExteNET trial is a double-blind, placebo-controlled, Phase III trial of neratinib versus placebo after adjuvant treatment with Herceptin in women with early stage HER2-positive breast cancer. More specifically, the ExteNET trial enrolled 2,840 patients in 41 countries with early stage HER2-positive breast cancer who had undergone surgery and adjuvant treatment with trastuzumab. After completion of adjuvant treatment with trastuzumab, patients were randomized to receive extended adjuvant treatment with either neratinib or placebo for a period of one year. Patients were then followed for recurrent disease, ductal carcinoma in situ (DCIS), or death for a period of two years after randomization in the trial.
The safety results of the study showed that the most frequently observed adverse event for the neratinib-treated patients was diarrhea, with approximately 39.9% of the neratinib-treated patients experiencing grade 3 or higher diarrhea (1 patient, 0.1%, had grade 4 diarrhea). Patients who received neratinib in this trial did not receive any prophylaxis with antidiarrheal agents to prevent the neratinib-related diarrhea. Puma’s previously reported clinical data from several trials have demonstrated that the use of high dose prophylactic loperamide may greatly reduce the rate of grade 3 diarrhea with neratinib, with grade 3 diarrhea rates ranging from 0-17% in studies in which high dose loperamide prophylaxis was used. Puma Biotechnology is currently conducting an international, open-label, Phase II study investigating the use of antidiarrheal prophylaxis with loperamide alone or with other agents in the prevention and reduction of neratinib-associated diarrhea and, more specifically, grade 3 diarrhea. The interim results of this trial (data cut-off of November 2016) showed that the incidence of grade 3 diarrhea for the total 135 patients who received the loperamide prophylaxis was 28.1% and that the incidence of grade 3 diarrhea was 15.0% for the 40 patients who received the combination of loperamide plus budesonide. In all of its current ongoing studies Puma is instituting the use of antidiarrheal prophylaxis for the first cycle of treatment in order to continue to reduce the neratinib-related diarrhea. See “–Safety Database” for additional information.
The primary endpoint of the trial was invasive disease-free survival (DFS). The results of the trial demonstrated that treatment with neratinib resulted in a 33% reduction of risk of invasive disease recurrence or death versus placebo (hazard ratio = 0.67, p = 0.009). The 2-year DFS rate for the neratinib arm was 93.9% and the 2-year DFS rate for the placebo arm was 91.6%. The secondary endpoint of the trial was disease-free survival including ductal carcinoma in situ (DFS-DCIS). The results of the trial demonstrated that treatment with neratinib resulted in a 37% reduction of risk of disease recurrence including DCIS or death versus placebo (hazard ratio = 0.63, p = 0.002). The 2-year DFS-DCIS rate for the neratinib arm was 93.9% and the 2-year DFS-DCIS rate for the placebo arm was 91.0%.
As an inclusion criteria for the ExteNET trial, patients needed to have tumors that were HER2-positive using local assessment. In addition, as a pre-defined subgroup in the trial, patients had centralized HER2 testing performed on their tumor as well. At the time the 2-year data was compiled, centralized HER2 testing had been performed on 1,704 (60%) of the patients in the ExteNET trial and further central testing on available samples was currently ongoing. For the 1,463 patients whose tumors were HER2-positive by central confirmation, the results of the trial demonstrated that treatment with neratinib resulted in a 49% reduction of risk of invasive disease recurrence or death versus placebo (hazard ratio = 0.51, p = 0.002). The 2-year DFS rate for the centrally confirmed patients in the neratinib arm was 94.7% and the 2-year DFS rate for the centrally confirmed patients in the placebo arm was 90.6%. For the patients in the trial whose tumors were HER2-positive by central confirmation, the results of the trial demonstrated that treatment with neratinib resulted in a 51% reduction of risk of disease recurrence including DCIS or death versus placebo (hazard ratio = 0.49, p < 0.001). The 2-year DFS-DCIS rate for the centrally confirmed patients in the neratinib arm was 94.7% and the 2-year DFS rate for centrally confirmed patients in the placebo arm was 90.2%.
For the pre-defined subgroup of patients with hormone receptor positive disease, the results of the trial demonstrated that treatment with neratinib resulted in a 49% reduction of risk of invasive disease recurrence or death versus placebo (hazard ratio = 0.51, p = 0.001). The 2-year DFS rate for the neratinib arm was 95.4% and the 2-year DFS rate for the placebo arm was 91.2%. For the patients in the trial whose tumors were HER2-positive by central confirmation, the results of the trial demonstrated that treatment with neratinib resulted in a 75% reduction of risk of invasive disease recurrence or death (hazard ratio = 0.25, p < 0.001). The 2-year DFS rate for the centrally confirmed patients in the neratinib arm was 97.0% and the 2-year DFS rate for centrally confirmed patients in the placebo arm was 88.4%.
Based on the results from the ExteNET trial, in June and July 2016, the company submitted an MAA with the EMA and filed an NDA with the FDA, respectively, for regulatory approval of neratinib in the extended adjuvant setting.
Five-Year ExteNET Data. In September 2017, the company presented updated data from the ExteNET trial at the European Society of Medical Oncology (ESMO) 2017 Congress in Madrid, Spain. The data represented a predefined 5-year invasive disease free survival (iDFS) analysis as a follow-up to the primary 2-year iDFS analysis of the Phase III ExteNet trial. The results of the trial demonstrated that after a median follow up of 5.2 years, treatment with neratinib resulted in a 27% reduction of risk of invasive disease recurrence or death versus placebo (hazard ratio = 0.73, p = 0.008). The 5-year iDFS rate for the neratinib arm was 90.2% and the 5-year iDFS rate for the placebo arm was 87.7%. The secondary endpoint of the trial was invasive disease free survival including ductal carcinoma in situ (iDFS-DCIS). The results of the trial demonstrated that treatment with neratinib resulted in a 29% reduction of risk of disease recurrence, including DCIS or death versus placebo (hazard ratio = 0.71, p = 0.004). The 5-year iDFS-DCIS rate for the neratinib arm was 89.7% and the 5-year iDFS-DCIS rate for the placebo arm was 86.8%.
For the pre-defined subgroup of patients with hormone receptor positive disease, the results of the trial demonstrated that treatment with neratinib resulted in a 40% reduction of risk of invasive disease recurrence or death versus placebo (hazard ratio = 0.60, p = 0.002). The 5-year iDFS rate for the neratinib arm was 91.2% and the 5-year iDFS rate for the placebo arm was 86.8%. For the pre-defined subgroup of patients with hormone receptor negative disease, the results of the trial demonstrated that treatment with neratinib resulted in a hazard ratio of 0.95 (p = 0.762).
The safety results were unchanged from the primary 2-year iDFS analysis of the study that showed the most frequently observed adverse event for the neratinib-treated patients was diarrhea, with approximately 39.9% of the neratinib-treated patients experiencing grade 3 or higher diarrhea (1 patient (0.1%) had grade 4 diarrhea). Patients who received neratinib in this trial did not receive any prophylaxis with antidiarrheal agents to prevent the neratinib-related diarrhea.
Neoadjuvant Breast Cancer
At the 2010 CTRC-AACR San Antonio Breast Cancer Symposium, the results of the Neoadjuvant Lapatinib and/or Trastuzumab Treatment Optimisation Study, or the Neo-ALTTO study, were presented. In this trial, patients with HER2-positive breast cancer were randomized to receive either the combination of paclitaxel plus trastuzumab, the combination of paclitaxel plus lapatinib or the combination of paclitaxel plus trastuzumab plus lapatinib, as a neoadjuvant (preoperative) therapy. The results of the trial demonstrated that patients who received the combination of paclitaxel plus trastuzumab demonstrated a pathological complete response rate, or pCR, of 27.6% in the breast and lymph nodes, the patients who received paclitaxel plus lapatinib had a pCR of 20.0% and the patients who received the combination of paclitaxel plus trastuzumab plus lapatinib had a pCR of 46.8%.
Also at the 2010 CTRC-AACR San Antonio Breast Cancer Symposium, the results of the Neo-Sphere study were presented. In this trial, patients with HER2-positive breast cancer were randomized to receive either the combination of docetaxel plus trastuzumab, the combination of docetaxel plus pertuzumab, the combination of trastuzumab plus pertuzumab or the combination of docetaxel plus trastuzumab plus pertuzumab, as a neoadjuvant (preoperative) therapy. The results of the trial demonstrated that the patients who received the combination of docetaxel plus trastuzumab had a pCR of 21.5%in the breast and lymph nodes, the patients who received docetaxel plus pertuzumab had a pCR of 17.7%, the patients who received pertuzumab plus trastuzumab had a pCR of 11.2% and the patients who received the combination of docetaxel plus trastuzumab plus pertuzumab had a pCR of 39.3%.
I-SPY 2 TRIAL. In 2010, the Foundation for the National Institutes of Health initiated the I-SPY 2 TRIAL (Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging And moLecular Analysis 2). The I-SPY 2 TRIAL is a randomized Phase II clinical trial for women with newly diagnosed Stage 2 or higher (tumor size at least 2.5 cm) breast cancer that addresses whether adding investigational drugs to standard chemotherapy in the neoadjuvant setting is better than standard chemotherapy. The primary endpoint was pCR in the breast and the lymph nodes at the time of surgery. The goal of the trial was to match investigational regimens with patient subsets on the basis of molecular characteristics, referred to as biomarker signatures, that benefit from the regimen.
The I-SPY 2 TRIAL involved an adaptive trial design based on Bayesian predictive probability that a regimen will be shown to be statistically superior to standard therapy in an equally randomized 300-patient confirmatory trial. Regimens that have a high Bayesian predictive probability of showing superiority in at least one of 10 predefined signatures graduate from the trial. Regimens are dropped for futility if they show a low predictive probability of showing superiority over standard therapy in all 10 signatures. A maximum total of 120 patients can be assigned to each experimental regimen. A regimen can graduate early and at any time after having 60 patients assigned to it.
In April 2014, the company announced the results for the neratinib-containing regimen of the I-SPY 2 TRIAL. The neratinib-containing regimen (neratinib plus paclitaxel followed by doxorubicin and cyclophosphamide) graduated from the I-SPY 2 TRIAL based on having a high probability of success in Phase III with a signature of HER2 positive/HR negative. In this group, treatment with the neratinib-containing regimen resulted in an estimated pCR rate of 55.6% compared to the control arm (standard neoadjuvant chemotherapy: paclitaxel in combination with trastuzumab followed by doxorubicin and cyclophosphamide), which had an estimated pCR rate of 32.6%. The Bayesian probability of superiority for the neratinib-containing regimen (compared to standard therapy) is 94.9%, which is analogous to a p-value of 0.051. In addition, the Bayesian predictive probability of showing statistical superiority in a 300-patient Phase III randomized trial of paclitaxel plus neratinib versus paclitaxel plus trastuzumab, both followed by doxorubicin/cyclophosphamide, is 79.1%.
For the 65 patients in the trial who were HER2 positive (including those who were either hormone receptor positive or negative), treatment with the neratinib-containing regimen resulted in an estimated pCR rate of 39.4% compared to the control arm, which demonstrated an estimated pCR rate of 22.8%. The Bayesian probability of superiority for the neratinib-containing regimen is 95.4%, which is analogous to a p-value of 0.046. In addition, the Bayesian predictive probability of showing statistical superiority in a 300-patient Phase III randomized trial of paclitaxel plus neratinib versus paclitaxel plus trastuzumab is 72.7%.
Patients in the I-SPY 2 TRIAL were screened using the MammaPrint 70-gene signature test to determine if they had a heightened risk of breast cancer recurrence. The median MammaPrint score from the patients in the previous I-SPY 1 TRIAL who fit the eligibility criteria for I-SPY 2 was used as a predefined stratification factor for the I-SPY 2 TRIAL. Patients in I-SPY 2 were stratified as either MammaPrint High (below the median from I-SPY 1) or MammaPrint Ultra High (above the median from I-SPY 1). For the 41 neratinib treated patients in the trial who were MammaPrint Ultra High (80.5% of whom were HER2 negative), treatment with the neratinib-containing regimen resulted in an estimated pCR rate of 47.5% compared to the control arm, which demonstrated an estimated pCR rate of 29.4%. The Bayesian probability of superiority for the neratinib-containing regimen is 93.3%, which is analogous to a p-value of 0.067. In addition, the Bayesian predictive probability of showing statistical superiority in a 300-patient Phase III randomized trial of paclitaxel plus neratinib versus paclitaxel alone for HER2-negative patients, or in combination with trastuzumab for the HER2-positive patients, is 71.8%.
The results of the I-SPY2 TRIAL with neratinib were published in The New England Journal of Medicine in July 2016.
FB-7 Trial. In 2010, Pfizer, in collaboration with the National Surgical Adjuvant Breast and Bowel Project, or NSABP, a clinical trials cooperative group supported by the National Cancer Institute, or NCI, initiated the FB-7 study to investigate the use of neratinib as a neoadjuvant therapy for newly diagnosed HER2-positive breast cancer. In this trial, a total of 126 patients are randomized to receive neratinib plus the chemotherapy drug paclitaxel or trastuzumab plus paclitaxel prior to having surgery to remove their tumors. The purpose of this study is to test whether adding neratinib to paclitaxel chemotherapy is better than trastuzumab plus paclitaxel chemotherapy before having surgery. This trial was modified in 2012 to include a third treatment arm where patients will receive the combination of neratinib plus trastuzumab plus paclitaxel prior to having surgery to remove their tumors.
Data from this trial were presented at the 2015 CTRC-AACR San Antonio Breast Cancer Symposium. Patients were randomly assigned to trastuzumab (T) or neratinib (N) or the combination (T+N) with weekly paclitaxel (P) followed by standard doxorubicin and cyclophosphamide chemotherapy (AC) administered prior to surgery. 126 U.S., Canadian, and European patients were randomly assigned to Arm 1 (T+P followed by AC), Arm 2 (N+P followed by AC) or Arm 3 (T+N+P followed by AC). The primary endpoint of the trial was pathological complete response rate (pCR) in the breast and lymph nodes. Tumor tissue was collected on patients at the time of diagnosis. This tissue will be analyzed for several biomarkers including AKT, cMET, EGFR, ESR-alpha, HER2, HER3, HER4, p95 HER2 and PI3K and intrinsic subtypes. A key secondary endpoint of this trial is the molecular and genetic correlates of response for each of these biomarkers.
For the intent-to-treat patient population (hormone receptor positive (HR+) and hormone receptor negative (HR-)), the pCR rate for Arm 1 was 38.1%, for Arm 2 was 33.3% and for Arm 3 was 50.0%. For the HR+ patients, the pCR rate for Arm 1 was 29.6%, for Arm 2 was 27.6% and for Arm 3 was 30.4%. For the HR- patients, the pCR rate for Arm 1 was 57.1%, for Arm 2 was 46.2% and for Arm 3 was 73.7%.
The most frequently observed severe adverse event in the two neratinib treated arms of the trial (Arm 2 and Arm 3) was diarrhea. In the first 19 patients treated in Arm 2 of the trial, high dose loperamide (16 mg per day initially) as primary prophylaxis was not given to prevent the neratinib-related diarrhea. In this subset of patients the grade 3 diarrhea rate was 42% (8/19). In the next 10 patients treated in Arm 2 and the first 20 patients treated in Arm 3, high dose primary prophylaxis (16 mg per day initially) with loperamide was given during the initial two weeks of the first cycle of treatment. Using two weeks of intensive loperamide prophylactically, the grade 3 diarrhea rate in Arm 2 was 30% (3/10) and the grade 3 diarrhea rate in Arm 3 was 35% (7/20). In the next 13 patients in Arm 2 and 22 patients in Arm 3, high dose prophylaxis (16 mg per day initially) was given for the entire first cycle of treatment (4 weeks). The grade 3 diarrhea rate was 15% (2/13) in Arm 2 and 23% (5/22) in Arm 3.
In December 2016, a biomarker analysis of the FB-7 trial was presented at the 2016 CTRC-AACR San Antonio Breast Cancer Symposium . Pre-treatment core biopsy samples (n=59) and post treatment surgical samples (n=17) were obtained from a subset of patients treated in the FB-7 trial. pCR data were available for 51 patients from the biomarker cohort. After excluding low tumor content non-evaluable samples, correlative biomarker analysis was performed in 42 patients.
Expression levels and the activation status of EGFR/HER2 signaling proteins were investigated. The results of the phosphorylated HER2 (phosphoHER2) showed that median levels of phosphoHER2 were higher in the patients who achieved a pCR with neratinib (n=7) than in the patients who did not achieve a pCR who received either trastuzumab (n=8, p=0.07) or the combination of trastuzumab plus neratinib (n=4, p=0.035). There was not a significant difference in the median levels of phosphoHER2 in the patients who achieved a pCR with neratinib (n=7), trastuzumab (n=8, p=0.16) or the combination of trastuzumab plus neratinib (n=4, p=0.10).
The truncated form of HER2 known as p95HER2 was measured by the proprietary assay of Pierian Bioscience. p95HER2 represents a truncated form of the HER2 receptor that lacks the extracellular trastuzumab binding domain. It is believed to represent a mechanism of trastuzumab resistance. Median p95HER2 levels were higher in samples from patients who achieved a pCR with neratinib than in the patients who did not achieve a pCR and who received either trastuzumab (p=0.027) or the combination of trastuzumab plus neratinib (p=0.009). There was not a significant difference in the median levels of p95HER2 in the patients who achieved a pCR with neratinib (n=7), trastuzumab (n=8, p=0.16) or the combination of trastuzumab plus neratinib (n=4, p=0.35).
The MammaPrint assay was performed on 59 samples to determine if there was any imbalance between arms. This assay is a genomic test that analyzes the activity of 70 genes and then calculates a recurrence score that is either low risk or high risk. The results of the MammaPrint showed that the patients in all three arms of the FB-7 trial were balanced with the median MammaPrint risk score being similar across arms. There were only three patients with a MammaPrint low score.
PB272 (neratinib, oral)—Metastatic Breast Cancer
Trials of Neratinib as a Single Agent. In 2009, Pfizer presented data at the CTRC-AACR San Antonio Breast Cancer Symposium from a Phase II trial of neratinib administered as a single agent to patients with HER2-positive metastatic breast cancer. Final results from this trial were published in the Journal of Clinical Oncology in March 2010.
The trial involved a total of 136 patients, 66 of whom had received prior treatment with trastuzumab and 70 of whom had not received prior treatment with trastuzumab. The results of the study showed that neratinib was reasonably well-tolerated among both the pretreated patients and the patients who had not received prior treatment with trastuzumab. Diarrhea was the most common side effect, but was manageable with antidiarrheal agents and dose modification. Efficacy results from the trial showed that the objective response rate was 24% for patients who had received prior trastuzumab treatment and 56% for patients with no prior trastuzumab treatment. Furthermore, the median PFS was 22.3 weeks for the patients who had received prior trastuzumab and 39.6 weeks for the patients who had not received prior trastuzumab.
Trials of Neratinib in Combination with Other Anti-Cancer Drugs. In November 2014, the company announced top line results from a Phase II clinical trial of neratinib for the treatment of first-line HER2-positive locally recurrent or metastatic breast cancer (NEfERTT trial). Data from this trial was presented at the American Society of Clinical Oncology (ASCO) 2015 Annual Meeting in June 2015. The NEfERTT trial was a randomized, two-arm Phase II trial of neratinib plus the anticancer drug paclitaxel versus trastuzumab (Herceptin) plus paclitaxel as a first-line treatment for HER2-positive locally recurrent or metastatic breast cancer. The trial enrolled 479 patients in 33 countries with locally recurrent or metastatic breast cancer who had not received prior anticancer therapy for locally recurrent or metastatic disease. Patients were randomized to receive first-line treatment with either paclitaxel plus neratinib or paclitaxel plus trastuzumab. The primary endpoint of the trial was progression free survival. The secondary endpoints of the study included objective response rate and the incidence of central nervous system (CNS) metastases, including brain metastases.
The results of the trial demonstrated that the progression free survival for the patients who received the combination of paclitaxel plus neratinib was 12.9 months and the progression free survival for the patients who received the combination of paclitaxel plus trastuzumab was 12.9 months (p=0.777). The objective response rate in the trial for the patients who received the combination of paclitaxel plus neratinib was 74.8% and the objective response rate for the patients who received the combination of paclitaxel plus trastuzumab was 77.6% (p=0.522). With respect to the incidence of central nervous system metastases (e.g., brain metastases), treatment with the combination of paclitaxel plus neratinib resulted in a 52% reduction in the incidence of CNS metastases compared to the incidence of CNS metastases in patients who received the combination of paclitaxel plus trastuzumab. Symptomatic or progressive CNS recurrences occurred in 20 patients (8.3%) in the neratinib-paclitaxel group and 41 patients (17.3%) in the trastuzumab-paclitaxel group (relative risk 0.48, p=0.002). The estimated Kaplan-Meier 2-year incidence of CNS recurrences was 16.3% in the neratinib-paclitaxel group and 31.2% in the trastuzumab-paclitaxel group (hazard ratio 0.45, p=0.004). These results reflect a statistically significant difference between the two treatment arms. The company believe that this represents the first randomized trial with a HER2 targeted agent that has shown a statistically significant reduction in the incidence of CNS metastases. The Phase II trial results were published online in the JAMA Oncology in April 2016.
Pfizer presented data from a Phase II trial at the 2010 CTRC-AACR San Antonio Breast Cancer Symposium, which evaluated the safety and efficacy of neratinib when given in combination with the anti-cancer drug vinorelbine in patients with HER2-positive metastatic breast cancer. In the 56 patients who had not been previously treated with the anti-HER2 therapy lapatinib, treatment with the combination of vinorelbine plus neratinib resulted in an overall response rate of 57% and PFS was 44.1 weeks. For those patients who had received prior treatment with lapatinib, the overall response rate was 50%. The combination of vinorelbine and neratinib was generally well tolerated.
Data from a third Phase II study, in which patients with confirmed HER2-positive metastatic breast cancer who had failed treatment with trastuzumab and taxane chemotherapy were given neratinib in combination with capecitabine, was presented at the 2011 CTRC-AACR San Antonio Breast Cancer Symposium. The results of the study showed that the combination of PB272 and capecitabine had acceptable tolerability. The efficacy results from the trial showed that for the 61 patients in the trial who had not been previously treated with the HER2 targeted anti-cancer drug lapatinib, there was an overall response rate of 64% and a clinical benefit rate of 72%. In addition, for the seven patients in the trial who had previously been treated with lapatinib, there was an overall response rate of 57% and a clinical benefit rate of 71%. The median PFS for patients who had not received prior treatment with lapatinib was 40.3 weeks and the median PFS for the patients who had received prior lapatinib treatment was 35.9 weeks.
In February 2013, the company reached agreement with the FDA under an SPA for its planned Phase III clinical trial of neratinib in patients with HER2-positive metastatic breast cancer who have failed two or more prior treatments (third-line disease). The SPA is a written agreement between it, as the trial’s sponsor, and the FDA regarding the design, endpoints, and planned statistical analysis of the Phase III trial with respect to the effectiveness of PB272 for the indication to be studied to support an NDA. The EMA has also provided follow-on SA, consistent with that of the FDA regarding its Phase III trial design and endpoints to be used and ability of such design to support the submission of an MAA in the EU.
Pursuant to the SPA and SA, the Phase III trial is designed as a randomized study of neratinib plus capecitabine versus lapatinib plus capecitabine in patients with third-line HER2-positive metastatic breast cancer. The trial is expected to enroll approximately 600 patients who will be randomized (1:1) to receive either PB272 plus capecitabine or lapatinib plus capecitabine. The trial will be conducted at approximately 250 sites in North America, Europe and Asia-Pacific. The agreed upon co-primary endpoints of the trial are PFS and overall survival. The company's plan is to use the PFS data from the trial as the basis for submission of an NDA, and its foreign equivalents for Accelerated/Conditional Approval for PB272 from the regulatory agencies. The company commenced patient enrollment in this Phase III trial in the second quarter of 2013. The company expect to report the top line data from this trial in 2018.
In 2010, Pfizer also initiated a Phase I/II trial of neratinib in combination with the anti-cancer drug temsirolimus, or Torisel, in patients with HER2-positive metastatic breast cancer who have failed multiple prior treatments. The trial was conducted as a Phase I/II trial of PB272 given in combination with the anticancer drug temsirolimus in patients with HER2-positive metastatic breast cancer. The Phase I portion of the trial, which was reported previously, determined that the maximum tolerated dose was 240 mg of neratinib daily with 8 mg of temsirolimus weekly and the dose limiting toxicity was diarrhea. The interim Phase II data was presented at the 2014 CTRC-AACR San Antonio Breast Cancer Symposium. The Phase II portion of the study was conducted in two cohorts. The first cohort, referred to as the Maximum Tolerated Dose (MTD) cohort, received 240 mg of neratinib daily with 8 mg of temsirolimus weekly. This cohort of patients received low dose loperamide (4 mg per day) prophylactically in order to reduce the neratinib-related diarrhea. The second cohort of patients, referred to as the Dose Escalation cohort (DE cohort), received 240 mg of neratinib daily and initially received 8 mg of temsirolimus weekly. This cohort of patients received high dose loperamide (16 mg per day initially) prophylactically in order to reduce the neratinib-related diarrhea. If patients in the DE cohort had no tolerability issues with the combination of neratinib and temsirolimus given at 8 mg per week during the first cycle of treatment, patients in this DE cohort were allowed to dose escalate the temsirolimus to 15 mg per week for the remainder of the study. Patients in both cohorts in the study received a median of 3 prior regimens in the metastatic setting (range 1-8 prior regimens) before entering the trial. The 37 patients in the MTD cohort were enrolled at 3 centers in the United States and the 45 patients in the DE cohort were enrolled at 8 centers in the United States, Europe and Asia. The interim safety results of the study showed that the most frequently observed adverse event for the patients who received the combination of neratinib plus temsirolimus was diarrhea. For the 37 patients in the MTD cohort, who received low dose loperamide prophylactically, 12 patients (32%) experienced grade 3 diarrhea. For the 41 patients in the DE cohort, who received high dose loperamide prophylactically and were allowed to dose escalate the temsirolimus dose, 7 patients (17%) reported grade 3 diarrhea. 4 (57%) of the 7 patients in the DE cohort who experienced grade 3 diarrhea were not compliant with the high dose loperamide prophylaxis. There were 4 patients in the DE cohort who did not yet have safety data reported and are therefore not included in the safety population. For the patients in the DE cohort, thus far 47% of the patients have been able to dose escalate temsirolimus from 8 mg per week to 15 mg per week. The interim efficacy results from the trial showed that for the 37 patients in the MTD cohort, 11 patients (30%) experienced a partial response (PR). The median duration of response for this cohort of patients was 3.0 months and the median progression-free survival was 4.8 months. For the 37 evaluable patients in the DE cohort, the efficacy results from the trial demonstrated that 11 patients (30%) experienced a PR.
Metastatic Breast Cancer with Brain Metastases
Approximately one-third of the patients with HER2-positive metastatic breast cancer develop metastases that spread to their brain. The current antibody-based treatments, including trastuzumab, pertuzumab and T-DM1, do not enter the brain and therefore are not believed to be effective in treating these patients. In a Phase II trial with lapatinib given as a single agent, lapatinib demonstrated a 6% objective response rate in the patients with HER2-positive metastatic breast cancer whose disease spread to their brain. In January 2012, a Phase II trial of neratinib as a single agent and in combination with the anticancer drug capecitabine in patients with HER2-positive metastatic breast cancer that has spread to their brain was initiated in conjunction with the Dana Farber Translational Breast Cancer Research Consortium. In June 2014, at the ASCO 2014 Annual Meeting, results from the first cohort (n=40) who were administered neratinib monotherapy was presented. The efficacy results from the first cohort of the trial showed that for the 40 evaluable patients, 3 (7.5%) patients experienced a PR, 4 (10%) patients experienced prolonged stable disease (SD) for greater than or equal to 6 months and 12 (30%) patients experienced SD for less than 6 months. The median progression-free survival of the 40 evaluable patients was seen to be 1.9 months and the median overall survival was seen to be 8.7 months.
In June 2017, the company presented additional data from this trial at the ASCO 2017 Annual Meeting. The multicenter Phase II clinical trial enrolled patients with HER2-positive metastatic breast cancer who have brain metasteses. The trial enrolled three cohorts of patients. Patients in the second cohort (n=5) represent patients who had brain metastases which were amenable to surgery and who were administered neratinib monotherapy prior to and after surgical resection. The third cohort (target enrollment=60) enrolled two sub-groups of patients (prior lapatinib-treated and no prior lapatinib) with progressive brain metastases who were administered neratinib in combination with the chemotherapy drug capecitabine. The oral presentation reflected only the patients in the third cohort of patients without prior lapatinib exposure (cohort 3A, n=37), who all had progressive brain metastases at the time of enrollment and who received the combination of capecitabine plus neratinib. Results from the second cohort and cohort 3B (prior lapatinib-treated) will be presented at a forthcoming medical meeting.
In cohort 3A, 30% of the patients had received prior craniotomy, 65% of the patients had received prior whole brain radiotherapy (WBRT), and 35% had received prior stereotactic radiosurgery (SRS) to the brain. No patients had received prior treatment with lapatinib.
The primary endpoint of the trial was central nervous system (CNS) Objective Response Rate according to a composite criteria that included volumetric brain MRI measurements, steroid use, neurological signs and symptoms, and RECIST evaluation for non-CNS sites. The secondary endpoint of the trial was CNS response by Response Assessment in Neuro-Oncology-Brain Metastases (RANO-BM) Criteria. The efficacy results from the trial showed that 49% of patients experienced a CNS Objective Response by the composite criteria. The results also showed that the CNS response rate using the RANO-BM criteria was 24%. The median time to CNS progression was 5.5 months and the median overall survival was 13.5 months, though 49% of patients remain alive and survival data are immature.
The results for cohort 3A showed that the most frequently observed severe adverse event for the 37 patients evaluable for safety was diarrhea. Patients received antidiarrheal prophylaxis consisting of high dose loperamide, given together with the combination of capecitabine plus neratinib for the first cycle of treatment in order to try to reduce the neratinib-related diarrhea. Among the 37 patients evaluable for safety, 32% of the patients had grade 3 diarrhea and 41% had grade 2 diarrhea.
Safety Database. The company's safety database includes over 3,000 patients who have been treated with neratinib. To date, the most significant grade 3 or higher adverse event associated with neratinib has been diarrhea, which occurs in approximately 30% of patients receiving the drug. Historically, once diarrhea occurred, patients were treated with loperamide and/or a reduction in the dose of neratinib. Puma Biotechnology has evaluated a prophylactic protocol pursuant to which a high dose of loperamide, approximately 16 mg, is given together with the initial dose of neratinib and then tapered down during the first cycle of treatment. The company plan to continue evaluating this protocol as the preliminary data has suggested that this prophylactic regimen significantly reduces the incidence of diarrhea with neratinib.
In February 2015, Puma initiated a Phase II open-label trial of neratinib monotherapy for one year in 120 patients with early HER2-positive breast cancer who have completed one year of adjuvant trastuzumab, or the CONTROL trial. The CONTROL trial is an international, open-label, Phase II study investigating the use of loperamide prophylaxis with or without other agents in the reduction of neratinib-associated diarrhea that has a primary endpoint of the incidence of grade 3 diarrhea. In the CONTROL trial, patients with HER2-positive early stage breast cancer who had completed trastuzumab-based adjuvant therapy received neratinib daily for a period of one year. The trial initially tested high dose loperamide prophylaxis given for the first 2 cycles (56 days) of treatment (12 mg on days 1-14, 8 mg on days 15-56 and as needed thereafter). In the original protocol, 4 mg loperamide is self-administered with the first dose of neratinib, followed by 2 mg loperamide every 4 hours for the first 3 days, reducing to 2 mg loperamide every 6 to 8 hours through the first 2 cycles of therapy. With Amendment 1 of the protocol, the loperamide dosing schedule was modified to simplify the regimen. Following Amendment 1 of the protocol, 4 mg loperamide is self-administered with the first dose of neratinib, followed by 4 mg loperamide three times a day for 2 weeks, followed by 4 mg loperamide twice daily through the first 2 cycles of therapy. After two cycles, patients do not take loperamide prophylactically but take it as needed throughout the remainder of the treatment duration if diarrhea occurs.
In December 2017, interim results from the CONTROL trial were presented at the 2017 CTRC-AACR San Antonio Breast Cancer Symposium. The CONTROL trial was then expanded to include two additional cohorts. One cohort received the combination of loperamide and budesonide and the other cohort received the combination of loperamide plus colestipol. Budesonide is a locally acting corticosteroid that the Company believes targets the inflammation identified in a preclinical model of neratinib-induced diarrhea and colestipol is a bile acid sequestrant that the Company believes targets potential bile acid malabsorption that could result from such inflammation.
The interim analysis of the trial presented in the poster included a total of 137 patients who received neratinib plus loperamide prophylaxis, 64 patients who received neratinib plus loperamide prophylaxis for 2 cycles and budesonide for 1 cycle, and 120 patients who received neratinib plus loperamide prophylaxis for 1 cycle and colestipol for 1 cycle. The results of the trial showed that the incidence of grade 3 diarrhea for the 137 patients who received the loperamide prophylaxis was 30.7%. For the 137 patients who received the loperamide prophylaxis, the median number of grade 3 diarrhea episodes per patient was 1 and the median cumulative duration of grade 3 diarrhea was 3 days. For the 137 patients who received loperamide prophylaxis, 20.4% discontinued neratinib due to diarrhea. For the 64 patients who received the combination of loperamide plus budesonide, the results of the trial showed that the incidence of grade 3 diarrhea was 26.6%. The median number of grade 3 diarrhea episodes per patient was 1 and the median cumulative duration of grade 3 diarrhea was 2 days. For the 64 patients who received loperamide plus budesonide prophylaxis, 10.9% discontinued neratinib due to diarrhea.
PB272 (neratinib, oral)—Other Potential Applications
Non-Small Cell Lung Cancer (NSCLC)
Approximately 2% to 4% of patients with NSCLC have a HER2 mutation in the kinase domain. This mutation is believed to narrow the ATP binding cleft, which results in increased tyrosine kinase activity. The mutation is also believed to result in increased PI3K activity and mTOR activation. Published data suggests that patients with HER2-mutated non-small cell lung cancer do not respond to platinum chemotherapy and do not respond to epidermal growth factor receptor inhibitors.
In September 2014, the company reported initial data from the ongoing, open label Phase II clinical trial of PB272 (neratinib) for the treatment of patients with NSCLC with HER2 mutations as a late-breaking oral presentation at the ESMO 2014 Congress. In the trial, patients with confirmed Stage IIIB or Stage IV NSCLC with documented somatic HER2 mutations were randomized to receive either oral neratinib monotherapy at a dose of 240 mg per day or the combination of oral neratinib (at a dose of 240 mg daily) with intravenous temsirolimus administered at a dose of 8 mg per week. In order to attempt to reduce the neratinib-related diarrhea, high-dose loperamide prophylaxis (Imodium) was given to all patients in both arms of the study beginning on day 1 of neratinib dosing. The data presented in the oral presentation involved a total of 27 patients who completed the first stage of the trial; 13 of these patients received neratinib monotherapy and 14 of these patients received the combination of neratinib plus temsirolimus. The results of the study showed that the combination of PB272 and temsirolimus had acceptable tolerability. Historically the most frequently seen adverse event associated with neratinib has been diarrhea. In the previous Phase I trial of neratinib plus temsirolimus (published in the Journal of Clinical Oncology in 2014) the diarrhea with neratinib was seen to be dose dependent and its incidence increased with increasing neratinib dosage. In that Phase I trial, grade 3 or higher diarrhea was seen in approximately 30% of the patients treated with doses of neratinib that were 200 mg or higher. In the Phase II study, all patients received high-dose loperamide in order to attempt to prevent or reduce the neratinib-related diarrhea. For the 13 patients enrolled in the neratinib monotherapy arm, 1 patient (8%) experienced grade 3 diarrhea, and for the 14 patients enrolled in the combination of neratinib plus temsirolimus arm, 2 patients (14%) experienced grade 3 diarrhea. There were no grade 4 diarrhea events seen in the trial. For the 3 patients in the study (1 in the monotherapy arm, 2 in the combination arm) who experienced grade 3 diarrhea, 2 of the 3 patients were not compliant with the loperamide prophylaxis regimen and were not taking loperamide at the onset of grade 3 diarrhea.
The efficacy results from the trial showed that for the 13 patients in the trial who received neratinib monotherapy, no patient experienced a partial response, 7 patients (54%) achieved stable disease and 4 patients (31%) achieved clinical benefit (defined as a partial response or stable disease for 12 or more weeks). For the 14 patients who received the combination of neratinib plus temsirolimus, 3 patients (21%) experienced a partial response, 11 patients (79%) experienced stable disease and 9 patients (64%) achieved clinical benefit. The median PFS of the neratinib monotherapy arm was 2.9 months and the median PFS of the arm that received neratinib plus temsirolimus was 4.0 months. Patients continue to be enrolled in the arm of the trial that is receiving the combination of neratinib plus temsirolimus.
HER2 Mutation-Positive Solid Tumors
Based on the results from the Cancer Genome Atlas Study, the company estimate that between 2% and 11% of each solid tumor has a mutation in HER2. In the United States, this includes new diagnoses of an estimated 7,000 – 7,500 patients with bladder cancer; 4,000 – 4,500 patients with colorectal cancer; 1,500 – 2,000 patients with glioblastoma; 1,000 patients with melanoma; 4,000 – 5,000 patients with prostate cancer; 1,000 patients with stomach cancer; and 1,000 – 2,000 patients with uterine cancer.
Basket Trial for HER2 Mutation-Positive Solid Tumors. In October 2013, the company announced that the company had initiated a Phase II clinical trial of neratinib as a single agent in patients with solid tumors that have an activating HER2 mutation (SUMMIT basket trial). The Phase II SUMMIT basket trial is an open-label, multicenter, multinational study to evaluate the safety and efficacy of PB272 administered daily to patients who have solid tumors with activating HER2 or HER3 mutations. The study initially included six cohorts (baskets) of patients, each of which will include one of the following cancers: ![]() bladder/urinary tract cancer; (ii) colorectal cancer; (iii) endometrial cancer; (iv) gastric/esophageal cancer; (v) ovarian cancer; and (vi) all other solid tumors (including prostate, melanoma and pancreatic cancer). Each basket will initially consist of seven patients. If a certain predetermined objective response rate is seen in the initial cohort of seven patients, the basket will be expanded to include a larger number of patients.
bladder/urinary tract cancer; (ii) colorectal cancer; (iii) endometrial cancer; (iv) gastric/esophageal cancer; (v) ovarian cancer; and (vi) all other solid tumors (including prostate, melanoma and pancreatic cancer). Each basket will initially consist of seven patients. If a certain predetermined objective response rate is seen in the initial cohort of seven patients, the basket will be expanded to include a larger number of patients.
In May 2014, the company announced that the company expanded the first cohort from the SUMMIT basket trial. The cohort that has been expanded includes patients with metastatic breast cancer that is not HER2 amplified or overexpressed (HER2 negative) and has a HER2 mutation. In April 2015, the company announced that the company expanded the cohort from the Phase II clinical trial of PB272 in patients with metastatic NSCLC that is not HER2 amplified or overexpressed (HER2 negative) and has a HER2 mutation. In December 2015, the company announced that the company expanded the cohort that includes patients with metastatic biliary duct (bile duct) cancer that is not HER2 amplified or overexpressed (HER2 negative) and has a HER2 mutation. In January 2017, the company announced that the company expanded the fourth cohort that includes patients with metastatic cervical cancer and whose tumors have a HER2 mutation. The cervical cancer patients initially entered the study in the “other solid tumors with a HER2 mutation” cohort and, due to the preliminary activity seen in the trial, the company expanded a separate cervical cancer cohort pursuant to the protocol for the trial. The expanded HER2-mutant cervical cancer cohort will now enroll a total of 18 patients.
HER2-Mutated, Non-Amplified Breast Cancer
A HER2 mutation in patients with HER2-negative breast cancer was identified as part of a study performed by the Cancer Genome Atlas Network and published in Cancer Discovery in December 2012. The company believe this mutation may occur in an estimated 2% of patients with breast cancer. Pre-clinical data from this publication demonstrated that neratinib was active in pre-clinical models of HER2-negative breast cancer that have this HER2 mutation and that neratinib has more anti-cancer activity than either trastuzumab or lapatinib in cells with this mutation. A Phase II trial of neratinib in HER2-negative breast cancer patients who have a HER2 mutation opened for enrollment in December 2012.
As stated above, in May 2014 the company expanded the first cohort from the SUMMIT basket trial. Interim results from this ongoing Phase II trial were presented at the 2017 American Association for Cancer Research Annual Meeting (AACR) during the plenary session of the meeting. All patients received loperamide (16 mg per day initially) prophylactically for the first cycle of treatment in order to reduce the neratinib-related diarrhea. Included in the presentation were data on 141 patients enrolled in the neratinib monotherapy arm of the trial, including 124 patients with HER2 mutations and 17 patients with HER3 mutations. This included patients with 21 unique tumor types, with the most common being breast, lung, bladder and colorectal cancer. There were also 30 distinct HER2 and 12 distinct HER3 mutations observed among these patients, with the most frequent HER2 variants involving S310, L755, A755_G776insYVMA and V777.
PB272 (neratinib, intravenous)
The company also plan to develop neratinib as an intravenously administered agent. The intravenous version of neratinib resulted in higher exposure levels of neratinib in pre-clinical models. The company believe this may result in higher blood levels of neratinib in patients, and may translate into enhanced efficacy. Puma Biotechnology is evaluating the intravenous formulation of neratinib and considering options relative to its development.
PB357
PB357 is an orally administered agent that is an irreversible TKI that blocks signal transduction through the epidermal growth factor receptors, HER1, HER2 and HER4. PB357 is structurally similar to PB272. Pfizer completed single-dose Phase I trials of PB357. Puma Biotechnology is evaluating PB357 and considering options relative to its development.
Clinical Testing of The company's Products in Development
Each of its products in development, and likely all future drug candidates the company in-license, will require extensive pre-clinical and clinical testing to determine the safety and efficacy of the product applications prior to seeking and obtaining regulatory approval. This process is expensive and time consuming. In completing these trials, Puma Biotechnology is dependent upon third-party consultants, consisting mainly of investigators and collaborators, who will conduct such trials.
The company and its third-party consultants conduct pre-clinical testing in accordance with Good Laboratory Practices, or GLP, and clinical testing in accordance with Good Clinical Practice standards, or GCP, which are international ethical and scientific quality standards utilized for pre-clinical and clinical testing, respectively. GCP is the standard for the design, conduct, performance, monitoring, auditing, recording, analysis and reporting of clinical trials and the FDA requires compliance with GCP regulations in the conduct of clinical trials. Additionally, its pre-clinical and clinical testing completed in the EU is conducted in accordance with applicable EU standards, such as the EU Clinical Trials Directive (Directive 2001/20/EC of April 4, 2001), or the EU Clinical Trials Directive, and the national laws of the 28 member states of the EU, or Member States, implementing its provisions.
Puma Biotechnology has entered into, and may enter into in the future, master service agreements with clinical research organizations, or CROs, with respect to initiating, managing and conducting the clinical trials of its products. These contracts contain standard terms for the type of services provided that contain cancellation clauses requiring between 30 and 45 days written notice and that obligate it to pay for any services previously rendered with prepaid, unused funds being returned to it.
Competition
The development and commercialization of new products to treat cancer is highly competitive, and the company face considerable competition from major pharmaceutical, biotechnology and specialty cancer companies. As a result, there are and will likely continue to be extensive research and substantial financial resources invested in the discovery and development of new cancer products. The company's competitors include, but are not limited to, Genentech, Novartis, Roche, Boehringer Ingelheim, Takeda, Daiichi Sankyo and Seattle Genetics. None of these companies are developing their drugs for the extended adjuvant treatment of early stage HER2-positive breast cancer that has been previously treated with a trastuzumab-containing regimen. All of these competitors are developing their drugs for the treatment of metastatic HER2-positive breast cancer. Puma Biotechnology is an early stage company with a limited history of operations, sales, marketing and commercial manufacturing. Many of its competitors have substantially more financial and technical resources than the company do. In addition, many of its competitors have more experience than Puma Biotechnology has in pre-clinical and clinical development, manufacturing, regulatory and global commercialization. Puma Biotechnology is also competing with academic institutions, governmental agencies and private organizations that are conducting research in the field of cancer.
The company anticipate that the company will face intense competition if Puma Biotechnology is able to commercialize additional product candidates. The company expect that its products under development and in clinical trials will address major markets within the cancer sector. The company's competition will be determined in part by the potential indications for which drugs are developed and ultimately approved by regulatory authorities. Additionally, the timing of market introduction of some of its potential products or of competitors’ products may be an important competitive factor. Accordingly, the speed with which the company can develop products, complete pre-clinical testing, clinical trials and approval processes, and supply commercial quantities to market are expected to be important competitive factors. The company expect that competition among products approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price, reimbursement and patent position.
Sales and Marketing
During 2017, in connection with FDA approval of NERLYNX, the company hired a U.S. specialty sales force of approximately 85 sales specialists who are focused on promoting NERLYNX to oncologists. This sales force is supported by an experienced sales leadership team comprised of regional sales managers, and its experienced commercial team comprised of experienced professionals in marketing, access and reimbursement, managed markets, marketing research, commercial operations, and sales force planning and management. In addition, its commercial infrastructure includes capabilities in manufacturing, medical affairs, quality control, and compliance.
The company launched NERLYNX in the United States in July 2017, and its focus is to establish NERLYNX as the first choice for extended adjuvant treatment of adult patients with early stage HER2-overexpressed/amplified breast cancer following adjuvant trastuzumab-based therapy.
In other markets outside of the United States in which NERLYNX may be approved, if any, the company may choose to commercialize NERLYNX independently or by establishing one or more strategic alliances such as the ones Puma Biotechnology has established for commercializing NERLYNX in South East Asia, beginning with Australia, Singapore, Malaysia, Brunei and New Zealand, Israel and in greater China, including mainland China, Taiwan, Hong Kong and Macau.
Intellectual Property and License Agreements
The company hold a worldwide exclusive license under its license agreement with Pfizer to four granted U.S. patents and nine pending U.S. patent applications, as well as foreign counterparts thereof, and other patent applications and patents claiming priority therefrom.
In the United States, Puma Biotechnology has a license to an issued patent, which currently will expire in 2025, for the composition of matter of neratinib, its lead compound. Puma Biotechnology has a license to an issued U.S. patent covering a family of compounds including neratinib, as well as equivalent patents in the EU and Japan, that currently expire in 2019. The company also have a license to an issued U.S. patent for the use of neratinib in the treatment of breast cancer, which currently expires in 2025, and an issued patent for the use of neratinib in the extended adjuvant treatment of early stage HER2 positive breast cancer that has previously been treated with a trastuzumab containing regimen that expires in 2030. In jurisdictions which permit such, the company will seek patent term extensions where possible for certain of its patents. The company plan to pursue additional patents in and outside the United States covering additional therapeutic uses and polymorphs of neratinib from these existing applications. In addition, the company will pursue patent protection for any new discoveries or inventions made in the course of its development of neratinib.
If the company obtain marketing approval for neratinib or other drug candidates in the United States or in certain jurisdictions outside the United States, the company may be eligible for regulatory protection, such as five years of new chemical entity exclusivity and, as mentioned below, up to five years of patent term extension potentially available in the United States under the Hatch-Waxman Amendments. In addition, eight to eleven years of data and marketing exclusivity potentially are available for new drugs in the European Union; up to five years of patent extension are potentially available in Europe (Supplemental Protection Certificate), and eight years of data exclusivity are potentially available in Japan. There can be no assurance that the company will qualify for any such regulatory exclusivity, or that any such exclusivity will prevent competitors from seeking approval solely on the basis of their own studies. See “Government Regulation” below.
The intellectual property portfolio that was licensed from Pfizer in 2011 when the company licensed neratinib included issued patents in a number of countries, including in Europe (EP 1848414), as well as pending patent applications in several countries, including the United States, relating to methods of treating gefitinib-and/or erlotinib-resistant cancer by administering an irreversible epidermal growth factor receptor inhibitor. More specifically, the patent that was issued in Europe in April 2011 included specific claims that included a pharmaceutical composition for use in treating cancer in a subject with a cancer having a mutation in epidermal growth factor receptor with a T790M mutation. On November 28, 2011, Boehringer Ingelheim International GmbH filed an opposition to this patent asking for this patent to be revoked. The Oral Proceedings of the European Patent Office were held in Munich, Germany on February 4, 2014. The decision of the European Patent Office was to uphold the granted claims of the European patent that relate to the T790M mutation without any modification. This included specific claims that include claims for the pharmaceutical composition comprising an irreversible epidermal growth factor receptor inhibitor for use in treating cancer in a subject having a T790M mutation, and claims for the pharmaceutical composition for use in the treatment of numerous cancers, including lung cancer and non-small cell lung cancer. In September 2015, the company were advised of the issuance by the United States Patent and Trademark Office of a Notice of Allowance for U.S. Patent Application 11/883,474 titled “Method for Treating Gefitinib Resistant Cancer.”
The company's goal is to obtain, maintain and enforce patent protection for its products, formulations, processes, methods and other proprietary technologies, preserve its trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. The company's policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for its current product candidates and any future product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the United States and abroad. However, even patent protection may not always provide it with complete protection against competitors who seek to circumvent its patents. See “Risk Factors—Risks Related to The company's Intellectual Property—The company's proprietary rights may not adequately protect its intellectual property and potential products, and if the company cannot obtain adequate protection of its intellectual property and potential products, the company may not be able to successfully market its potential products.”
The company depend upon the skills, knowledge and experience of its scientific and technical personnel, as well as that of its advisors, consultants and other contractors, none of which is patentable. To help protect its proprietary know-how, which is not patentable, and inventions for which patents may be difficult to obtain or enforce, the company rely on trade secret protection and confidentiality agreements to protect its interests. To this end, the company require all of its employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to it of the ideas, developments, discoveries and inventions important to its business.
In-License Agreement
In August 2011, Former Puma entered into an agreement pursuant to which Pfizer, or Licensor, agreed to grant to Former Puma a worldwide license for the development, manufacture and commercialization of neratinib (oral), neratinib (intravenous), PB357, and certain related compounds. Pursuant to the terms of the agreement, the license would not become effective until Former Puma closed a capital raising transaction in which it raised at least $25 million in aggregate net proceeds and had a net worth of at least $22.5 million. Upon the closing of the financing that preceded the Merger, this condition was satisfied.
The company assumed the license agreement, in accordance with its terms, in the Merger. The license is exclusive with respect to certain patent rights owned or licensed by Pfizer. Under the license agreement, the Licensor is obligated to transfer to it certain information, records, regulatory filings, materials and inventory controlled by the Licensor and relating to or useful for developing these compounds and to continue to conduct certain ongoing clinical studies until a certain time. After that time, Puma Biotechnology is obligated to continue such studies pursuant to an approved development plan, including after the license agreement terminates for reasons unrelated to the Licensor’s breach of the license agreement, subject to certain specified exceptions. Puma Biotechnology is also obligated to commence a new clinical trial for a product containing one of these compounds within a specified period of time and use commercially reasonable efforts to complete such trial and achieve certain milestones as provided in a development plan. If certain of its out-of-pocket costs in completing such studies exceed a mutually agreed amount, the Licensor will pay for certain additional out-of-pocket costs to complete such studies. The company must use commercially reasonable efforts to develop and commercialize products containing these compounds in specified major-market countries and other countries in which the company believe it is commercially reasonable to develop and commercialize such products.
As consideration for the license, Puma Biotechnology is required to make payments totaling $187.5 million upon the achievement of certain milestones if all such milestones are achieved. In connection with the FDA approval of NERLYNX in July 2017, the company triggered a one-time milestone payment.
The license agreement originally stipulated that should the company commercialize any of the compounds licensed from the Licensor or any products containing any of these compounds, the company will be obligated to pay to the Licensor incremental annual royalties between approximately 10% and 20% of net sales of all such products, subject, in some circumstances, to certain reductions.
In July 2014, the Company signed an amendment to the license agreement with the Licensor. The amendment to the license agreement provides that the Company would be solely responsible for the expenses incurred or accrued in conducting the ongoing legacy clinical trials after December 31, 2013. These costs were previously the responsibility of the Licensor.
In addition, under the amended agreement, annual royalties to be paid on net sales of licensed products were reduced from a tiered royalty rate structure ranging between 10% to 20% to a fixed rate in the low to mid-teens. The Licensor and the Company have agreed to continue to cooperate to effect the transfer to the Company of certain records, regulatory filings, materials and inventory controlled by the Licensor as promptly as reasonably practicable.
The company's royalty obligation continues, on a product-by-product and country-by-country basis, until the later of ![]() the last to expire valid claim of a licensed patent covering the applicable licensed product in such country, or (ii) the earlier of generic competition for such licensed product reaching a certain level of sales in such country or expiration of a certain time period after first commercial sale of such licensed product in such country. When sublicensing the rights granted to it under the license agreement with the Licensor to a third party, the same milestone and royalty payments are required. The company can terminate the license agreement at will at any time after April 4, 2013, or for safety concerns, in each case upon specified advance notice. Each party may terminate the license agreement if the other party fails to cure any breach of a material obligation by such other party within a specified time period. The Licensor may terminate the license agreement in the event of its bankruptcy, receivership, insolvency or similar proceeding. The license agreement contains other customary clauses and terms as are common in similar agreements in the industry.
the last to expire valid claim of a licensed patent covering the applicable licensed product in such country, or (ii) the earlier of generic competition for such licensed product reaching a certain level of sales in such country or expiration of a certain time period after first commercial sale of such licensed product in such country. When sublicensing the rights granted to it under the license agreement with the Licensor to a third party, the same milestone and royalty payments are required. The company can terminate the license agreement at will at any time after April 4, 2013, or for safety concerns, in each case upon specified advance notice. Each party may terminate the license agreement if the other party fails to cure any breach of a material obligation by such other party within a specified time period. The Licensor may terminate the license agreement in the event of its bankruptcy, receivership, insolvency or similar proceeding. The license agreement contains other customary clauses and terms as are common in similar agreements in the industry.
Out-License Agreements
In November 2017 and January 2018, the company entered into three exclusive license agreements with STA, Medison and CANbridge granting them the right to seek the regulatory approval and, if approved, commercialize NERLYNX in South East Asia, Israel and in greater China, respectively. Under each of the agreements, Puma Biotechnology is entitled to upfront and milestone payments throughout the term of the applicable agreement, as well as significant double-digit royalties calculated as a percentage of net sales of the licensed products in the respective territory. The description below provides a summary of its agreements with Specialised Therapeutics and CANbridge.
Specialised Therapeutics Agreement
On November 20, 2017, the company entered into a license agreement, or the Specialised Therapeutics Agreement, with STA. Pursuant to the Specialised Therapeutics Agreement, the company granted to STA, under certain of its intellectual property rights relating to neratinib, an exclusive (including with respect to it and its affiliates), sublicensable license to commercialize any pharmaceutical product containing neratinib in finished form, or, for purposes of this description, the Licensed Product, for the extended adjuvant treatment of adult patients with early stage HER2-positive breast cancer and HER2-positive metastatic breast cancer in Australia, Brunei, Cambodia, Indonesia, Laos, Malaysia, Myanmar, New Zealand, Papua New Guinea, Philippines, Singapore, Thailand, Timor-Leste and Vietnam, or the STA Territory.
The Specialised Therapeutics Agreement sets forth the parties’ respective obligations with respect to the development, commercialization and supply of the Licensed Product. Within the STA Territory, STA will be generally responsible for regulatory and commercialization activities, and the company will be solely responsible for the manufacturing and supply of the Licensed Product under a supply agreement that will be entered into between the parties.
Pursuant to the Specialised Therapeutics Agreement, Puma Biotechnology is entitled to upfront and other milestone payments of up to $4.5 million, payable upon achievement of the milestone events specified in the Specialised Therapeutics Agreement. Furthermore, Puma Biotechnology is entitled to receive significant double digit royalties calculated as a percentage of net sales of Licensed Products in the STA Territory.
The term of the Specialised Therapeutics Agreement continues, on a country-by-country basis, until the later of ![]() the expiration or abandonment of the last patent covering the Licensed Product or (ii) the earlier of (a) the date upon which sales of generic versions of Licensed Product reach a specified level in such country, or (b) the tenth anniversary of the first commercial sale of the Licensed Product in such country. The Specialised Therapeutics Agreement may be terminated by either party if the other party commits a material breach, subject to a customary cure period, or if the other party is insolvent. The Specialised Therapeutics Agreement will also terminate upon the termination of the supply agreement for Licensed Products between the parties.
the expiration or abandonment of the last patent covering the Licensed Product or (ii) the earlier of (a) the date upon which sales of generic versions of Licensed Product reach a specified level in such country, or (b) the tenth anniversary of the first commercial sale of the Licensed Product in such country. The Specialised Therapeutics Agreement may be terminated by either party if the other party commits a material breach, subject to a customary cure period, or if the other party is insolvent. The Specialised Therapeutics Agreement will also terminate upon the termination of the supply agreement for Licensed Products between the parties.
CANbridge Agreement
On January 30, 2018, the company entered into an exclusive license agreement, or the CANbridge Agreement, with CANbridge. Pursuant to the CANbridge Agreement, the company granted to CANbridge, under certain of its intellectual property rights relating to neratinib, an exclusive, sublicensable (under certain circumstances) license to develop and commercialize any pharmaceutical product containing neratinib, or, for purposes of this description, the Licensed Product, for the treatment of human disease, or for purposes of this description, the Field, in the People’s Republic of China, or the CANbridge Territory, including mainland China, Hong Kong, Macao, and Taiwan, or, each, a CANbridge Region.
The CANbridge Agreement sets forth the parties’ respective obligations with respect to the development, commercialization and supply of the Licensed Product. CANbridge will, at its expense, develop the Licensed Product for the purpose of obtaining regulatory approval in the Field and in the CANbridge Territory, subject to its approval of certain aspects of clinical studies conducted by CANbridge. Within the CANbridge Territory, CANbridge will be solely responsible, at its expense, for regulatory and commercialization activities. The company will be solely responsible, subject to certain exceptions, for the manufacturing and supply of the Licensed Product under a supply agreement that will be entered into between the parties.
Pursuant to the CANbridge Agreement, the company will receive an upfront payment of $30 million and potentially receive regulatory milestone payments totaling up to $40 million and sales-based milestone payments totaling up to $185 million. In addition, Puma Biotechnology is entitled to receive significant double-digit royalties calculated as a percentage of net sales of the Licensed Products in the CANbridge Territory.
The term of the CANbridge Agreement continues, on a CANbridge Region-by-CANbridge Region basis, until ![]() the later of the expiration or abandonment of the last licensed patent covering the Licensed Product in such CANbridge Region or (ii) the earlier of the date upon which sales of generic versions of the Licensed Product reach a specified level in such CANbridge Region, or
the later of the expiration or abandonment of the last licensed patent covering the Licensed Product in such CANbridge Region or (ii) the earlier of the date upon which sales of generic versions of the Licensed Product reach a specified level in such CANbridge Region, or ![]() the tenth anniversary of the first commercial sale of the Licensed Product in such CANbridge Region. The CANbridge Agreement may be terminated by either party if the other party commits a material breach, subject to a customary cure period, or if the other party is insolvent; provided that if CANbridge materially breaches its development or commercialization obligations in a particular CANbridge Region, the company may terminate the CANbridge Agreement solely with respect to such CANbridge Region. CANbridge may terminate the agreement at its convenience.
the tenth anniversary of the first commercial sale of the Licensed Product in such CANbridge Region. The CANbridge Agreement may be terminated by either party if the other party commits a material breach, subject to a customary cure period, or if the other party is insolvent; provided that if CANbridge materially breaches its development or commercialization obligations in a particular CANbridge Region, the company may terminate the CANbridge Agreement solely with respect to such CANbridge Region. CANbridge may terminate the agreement at its convenience.




