Radius Health
Overview
Radius Health (RDUS) is a science-driven fully integrated biopharmaceutical company that is committed to developing and commercializing innovative endocrine therapeutics in the areas of osteoporosis and oncology. In April 2017, its first commercial product, TYMLOSTM (abaloparatide) injection, was approved by the U.S. Food and Drug Administration ("FDA") for the treatment of postmenopausal women with osteoporosis at high risk for fracture defined as history of osteoporotic fracture, multiple risk factors for fracture, or patients who have failed or are intolerant to other available osteoporosis therapy. In May 2017, the company commenced U.S. commercial sales of TYMLOS and, as of February 2018, TYMLOS was available and covered for approximately 259 million U.S. insured lives, representing approximately 86% of U.S. insured lives. In May 2017, the company also announced positive top-line results from its completed 24-month ACTIVExtend clinical trial for TYMLOS, which met all of its primary and secondary endpoints. The company submitted a labeling supplement to the FDA in connection with the results from its ACTIVExtend trial in December 2017. In July 2017, the company entered into a license and development agreement with Teijin Limited (“Teijin”) for abaloparatide for subcutaneous injection (“abaloparatide-SC”) in Japan. Under this agreement, Radius Health is entitled to receive milestone payments upon the achievement of certain regulatory and sales milestones and a fixed low double-digit royalty based on net sales of abaloparatide-SC in Japan during the royalty term, and Radius Health has an option to negotiate for a co-promotion agreement with Teijin for abaloparatide-SC in Japan. The company's European Marketing Authorisation Application (“MAA”) for abaloparatide-SC is under review by the Committee for Medicinal Products for Human Use (“CHMP”) of the European Medicines Agency (“EMA”) and the company expect an opinion from the CHMP regarding the MAA during the first half of 2018. In the first quarter of 2018, the company expect to initiate a clinical trial in men with osteoporosis which, if successful, will form the basis of a supplemental new drug application ("NDA") seeking to expand the use of TYMLOS to treat men with osteoporosis at high risk for fracture. In the first half of 2018, the company plan to initiate a bone histomorphometry study, which would enroll approximately 25 postmenopausal women with osteoporosis to evaluate the early effects of TYMLOS on tissue-based bone remodeling and structural indices.1
Radius Health is developing an abaloparatide transdermal patch, or abaloparatide-patch, for potential use in the treatment of postmenopausal women with osteoporosis. In January 2018, the company met with the FDA and gained alignment with the agency on a single, pivotal BMD non-inferiority bridging study to support an NDA submission. The FDA agreed that, depending on the study results, a randomized, open label, active-controlled, non-inferiority Phase 3 study of up to 500 patients with postmenopausal osteoporosis at high risk of fracture would be sufficient to gain approval for abaloparatide-patch. The FDA confirmed that the primary endpoint will be change in lumbar spine BMD at 12 months and that the non-inferiority margin must preserve 75% of the active control (abaloparatide-SC) based on the lower bound of the 95% confidence interval. The company expect to initiate this pivotal study in mid-2019 and to complete it in 2020. In February 2018, the company entered into a scale-up and commercial supply agreement with 3M Company pursuant to which 3M has agreed to exclusively manufacture Phase 3 and global commercial supplies of abaloparatide-patch.
Radius Health is also developing its investigational product candidate, elacestrant (RAD1901), a selective estrogen receptor degrader (“SERD”), for potential use in the treatment of hormone-receptor positive breast cancer. Radius Health has completed enrollment in its ongoing dose escalation Part A, and dose expansion Part B and C, and in the 18F fluoroestradiol positron emission tomography (“FES-PET”) imaging Phase 1 studies of elacestrant in advanced metastatic breast cancer. In October 2017, the FDA granted Fast Track designation for its elacestrant breast cancer program. Based on feedback from the EMA and the FDA, the company now intend to conduct a single, randomized, controlled Phase 2 trial of elacestrant as a third-line monotherapy in approximately 300 patients with ER+/HER2- advanced/metastatic breast cancer. Patients in the study would be randomized to receive either elacestrant or the investigator’s choice of an approved hormonal agent and the primary endpoint of the study will be progression-free survival (“PFS”). The study would also include a planned interim PFS analysis. The company believe that, depending on results, this single trial would support applications for global marketing approvals for elacestrant as a third-line monotherapy. In addition, depending on results of the interim analysis, the Company could seek accelerated approval for elacestrant in the United States. The company will provide further study details when the Phase 2 study is started, which the company expect will be in the second half of 2018.
Radius Health is developing its internally discovered investigational product candidate, RAD140, a non-steroidal selective androgen receptor modulator ("SARM") for potential use in the treatment of hormone-receptor positive breast cancer. In September 2017, the company initiated a Phase 1 study of RAD140 in patients with locally advanced or metastatic breast cancer. The company expect to provide an update on its RAD140 development program by the end of 2018.
Marketed Product and Investigational Product Candidates
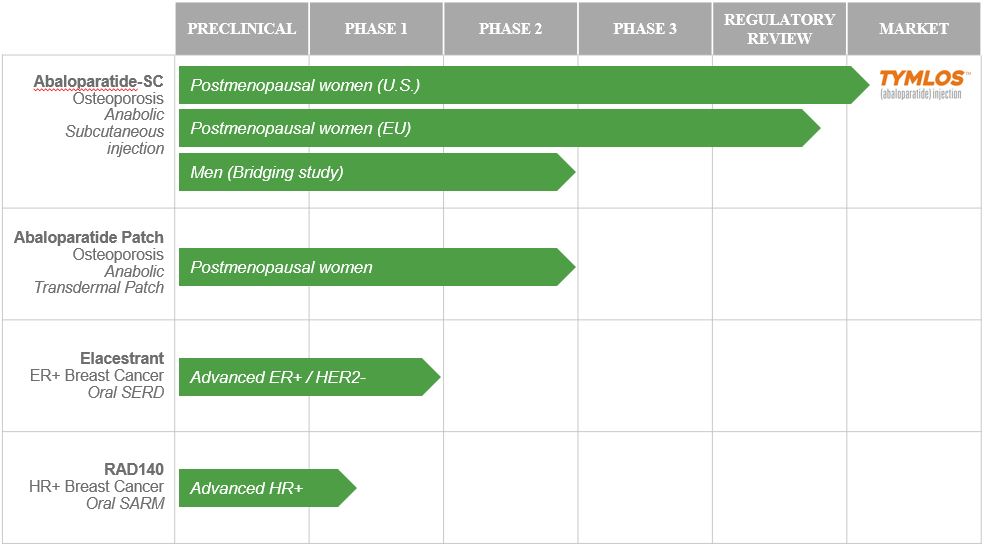
The success of its business is primarily dependent upon its ability to commercialize TYMLOS and to develop and commercialize its current and future product candidates. The following table identifies its commercial product, TYMLOS, and the investigational product candidates in its current product portfolio, their potential indication and stage of development. The company hold worldwide commercialization rights for all these product candidates, excluding abaloparatide-SC, for which the company hold worldwide commercialization rights, except for Japan, where Radius Health has an option to negotiate a co-promotion agreement with Teijin.
Abaloparatide
In April 2017, the FDA approved TYMLOS for the treatment of postmenopausal women with osteoporosis at high risk for fracture defined as history of osteoporotic fracture, multiple risk factors for fracture, or patients who have failed or are intolerant to other available osteoporosis therapy. Radius Health is developing two formulations of abaloparatide: abaloparatide-SC, an injectable subcutaneous formulation of abaloparatide, and abaloparatide-patch, a short-wear-time patch formulation of abaloparatide.
TYMLOS (Abaloparatide-SC)
TYMLOS was approved in the United States in April 2017 for the treatment of postmenopausal women with osteoporosis at high risk for fracture. The efficacy of TYMLOS for the treatment of postmenopausal women with osteoporosis was evaluated in an 18-month, randomized, multicenter, double-blind, placebo controlled clinical trial. The study also had an open label teriparatide arm. TYMLOS resulted in a significant reduction in the incidence of new vertebral fractures compared to placebo at 18 months (0.6% TYMLOS compared to 4.2% placebo). The relative risk reduction in new vertebral fractures at 18 months compared to placebo was 86% (p <0.001) for TYMLOS and 80% (p <0.001) for teriparatide. TYMLOS also resulted in a significant reduction in the incidence of nonvertebral fractures at the end of the 18 months of treatment plus one month follow-up where no drug was administered (2.7% for TYMLOS compared to 4.7% for placebo). The relative risk reduction, compared to placebo, in nonvertebral fractures for TYMLOS was 43% (p=0.049) and 28% for teriparatide (p=0.22). There was a 43% (p=0.02) risk reduction in clinical fractures for TYMLOS and a 29% (p=0.11) for teriparatide; a 70% (p<0.001) risk reduction in major osteoporotic fractures for TYMLOS, and 33% (p=0.14) for teriparatide, compared with patients who received placebo. For major osteoporotic fractures, there was a 55% (p=0.03) risk reduction for TYMLOS-treated patients compared to teriparatide.
The first commercial sales of TYMLOS in the United States occurred in May 2017 and Radius Health is commercializing TYMLOS in the United States through its commercial organization. Radius Health has built a distribution network for TYMLOS in the United States, comprised of well-established distributors and specialty pharmacies. Under its distribution model, both the distributors and specialty pharmacies take physical delivery of TYMLOS and the specialty pharmacies dispense TYMLOS directly to patients. As of February 2018, TYMLOS was available and covered for approximately 259 million U.S. insured lives, representing approximately 86% of U.S. insured lives. The company hold worldwide commercialization rights to abaloparatide-SC, except for Japan, where Radius Health has an option to negotiate a co-promotion agreement with Teijin for abaloparatide-SC.
The combined 25-month fracture data from its Phase 3 clinical trial program for TYMLOS formed the basis of its regulatory submissions in the United States and Europe. In November 2015, the company submitted an MAA for abaloparatide-SC to the EMA, which was validated and is currently undergoing active regulatory assessment by the CHMP. In December 2017, the CHMP issued a third Day-180 List of Outstanding Issues. As part of its on-going risk-benefit assessment, the CHMP informed the Company that it intends to refer the MAA to a scientific advisory group for additional advice. The company expect that the CHMP may adopt an opinion regarding its MAA during the first half of 2018. The company intend to enter into a collaboration for the commercialization of abaloparatide-SC outside of the United States and Japan.
In May 2017, the company announced positive top-line results from the completed 24-month ACTIVExtend clinical trial of TYMLOS, which met all of its primary and secondary endpoints. In ACTIVExtend, patients who had completed 18 months of TYMLOS injections or placebo in the ACTIVE Phase 3 trial were transitioned to receive 24 additional months of open-label alendronate. For the subset of ACTIVE trial patients (n=1139) that enrolled in the ACTIVExtend trial, the previous TYMLOS-treated patients had a significant 84% relative risk reduction (p<0.0001) in the incidence of new vertebral fractures compared with patients who received placebo followed by alendronate. They also demonstrated a 39% risk reduction in nonvertebral fractures (p=0.038), a 34% risk reduction clinical fractures (p=0.045) and a 50% risk reduction in major osteoporotic fractures (p=0.011) compared with patients who received placebo followed by alendronate. At the 43-month timepoint, for all patients (n=1645) that enrolled in the ACTIVE trial, TYMLOS-treated patients had a statistically significant risk reduction in new vertebral fractures (p<0.0001), nonvertebral fractures (p=0.038), clinical fractures (p=0.045), and major osteoporotic fractures (p<0.001), compared with patients who received placebo followed by alendronate. While not a pre-specified endpoint, there was also a statistically significant risk reduction in hip fractures (p=0.027) at the 43-month time point in the TYMLOS-treated patients, compared with patients who received placebo followed by alendronate. The adverse events reported during the alendronate treatment period were similar between the previous TYMLOS-treated patients and the previous placebo group. The incidences of cardiovascular adverse events including serious adverse events were similar between groups. There have been no cases of osteonecrosis of the jaw or atypical femoral fracture in the entire TYMLOS development program. The results from the completed ACTIVExtend trial were presented at a major scientific meeting in September 2017 and the company submitted a labeling supplement in connection with this data to the FDA in December 2017.
In July 2017, the company entered into a license and development agreement with Teijin for abaloparatide-SC in Japan. Pursuant to the agreement, the company may receive additional milestone payments upon the achievement of certain regulatory and sales milestones, and a fixed low double-digit royalty based on net sales of abaloparatide-SC in Japan during the royalty term. In addition, Radius Health has an option to negotiate for a co-promotion agreement with Teijin for abaloparatide-SC in Japan.
In late 2017, the company gained agreement with the FDA on the design of a clinical trial in men with osteoporosis which, if successful, will form the basis of a supplemental NDA seeking to expand the use of TYMLOS to treat men with osteoporosis at high risk for fracture. The study will be a randomized, double-blind, placebo-controlled trial that will enroll approximately 225 men with osteoporosis. The primary endpoint is change in spine BMD at 12 months compared with placebo. In previous clinical trials, TYMLOS has demonstrated increases in BMD in postmenopausal women. The study will include specialized high-resolution imaging of bone structure in a subset of the study participants. The company expect to initiate the trial in the first quarter of 2018.
In the first half of 2018, the company plan to initiate a bone histomorphometry study, which would enroll approximately 25 postmenopausal women with osteoporosis to evaluate the early effects of TYMLOS on tissue-based bone remodeling and structural indices.
Abaloparatide-patch
Radius Health is also developing abaloparatide-patch, based on 3M’s patented Microstructured Transdermal System technology, for potential use as a short wear-time transdermal patch. The company hold worldwide commercialization rights to the abaloparatide-patch technology and Radius Health is developing abaloparatide-patch toward future global regulatory submissions to build upon the potential success of TYMLOS. The company's development strategy for abaloparatide patch is to bridge to the established efficacy and safety of its approved abaloparatide-SC formulation.
The company commenced a human replicative clinical evaluation of the optimized abaloparatide-patch in December 2015, with the goal of achieving comparability to abaloparatide-SC. In September 2016, the company presented results from this evaluation of the first and second abaloparatide-patch prototypes, demonstrating that formulation technology can modify the pharmacokinetic profile of abaloparatide, including Tmax, half-life (“T1/2”), and area under the curve (“AUC”). In March 2018, the company announced that through further optimization the company had achieved comparability to the abaloparatide-SC profile with a third prototype (the “current abaloparatide-patch”). The current abaloparatide-patch optimized the drug-device combination through process improvements, a finalized formulation, selection of a dose (300 µg), and the introduction of a new clinical applicator. Together these changes, which were designed to improve the ease of use and patient experience, resulted in an increased half-life and AUC (915 pg.hr/ml for the current abaloparatide-patch, compared to 242 pg.hr/ml for the first patch prototype, 645 pg.hr/ml for the second patch prototype, and 936 pg.hr/ml for abaloparatide-SC).
In January 2018, the company met with the FDA to align on a regulatory and development path for registration of abaloparatide-patch. The company gained alignment with the agency on a single, pivotal BMD non-inferiority bridging study to support an NDA submission. The FDA agreed that, depending on the study results, a randomized, open label, active-controlled, non-inferiority Phase 3 study of up to 500 patients with postmenopausal osteoporosis at high risk of fracture would be sufficient to gain approval for abaloparatide-patch. The FDA confirmed that the primary endpoint will be change in lumbar spine BMD at 12 months and that the non-inferiority margin must preserve 75% of the active control (abaloparatide-SC) based on the lower bound of the 95% confidence interval. The company expect to initiate this pivotal study in mid-2019 and to complete it in 2020. On February 27, 2018, the company entered into a scale-up and commercial supply agreement with 3M Company pursuant to which 3M has agreed to exclusively manufacture Phase 3 and global commercial supplies of abaloparatide-patch.
Elacestrant (RAD1901)
Elacestrant is a SERD that Radius Health is evaluating for potential use as an oral treatment for hormone-receptor positive breast cancer. The company hold worldwide commercialization rights to elacestrant. Elacestrant is currently being investigated in women with advanced estrogen receptor positive (“ER-positive”) and human epidermal growth factor receptor 2-negative (“HER2-negative”) breast cancer, the most common subtype of the disease. Studies completed to date indicate that the compound has the potential for use as a single agent, or in combination with other therapies for the treatment of breast cancer. To date, no dose limiting toxicities have been reported in the elacestrant program. The company believe that, subject to successful development, regulatory review and approval, elacestrant could have the potential to offer the following advantages compared to current standard of care treatments for ER-positive and HER2-negative breast cancer:
- ability to degrade the estrogen receptor;
- favorable efficacy and tolerability profile;
- ability to effectively combine with other agents;
- treatment of hormone-resistant breast cancers; and
- once a day oral administration.
Radius Health has completed enrollment in its elacestrant FES-PET imaging study and dose-escalation Part A and expansion study parts B and C Phase 1 breast cancer trials. In June 2017, the company discussed the data from these ongoing Phase 1 studies with the FDA to gain alignment on defining the next steps for its elacestrant breast cancer program, including the design of a Phase 2 trial. In this meeting, the FDA agreed that a single-arm monotherapy Phase 2 study of up to 200 patients, could be appropriate with the primary endpoint being ORR, coupled with DOR. Depending on the study results, which must demonstrate an improvement over then available therapies, this study could be considered a pivotal study for accelerated approval as long as a confirmatory study is ongoing at the time of its NDA submission. In October 2017, the FDA granted Fast Track designation for its elacestrant breast cancer program. Fast Track is a process the FDA designed to facilitate the development and expedite the review of new therapies to treat serious conditions and fill unmet medical needs.
In February 2018, the company received scientific advice from the European Medicines Agency (“EMA”) regarding a potential single-arm monotherapy Phase 2 trial of elacestrant in patients with ER+, HER2- advanced or metastatic breast cancer. In addition, the company had a further meeting in February 2018 with the FDA regarding the registrational pathway for elacestrant at which the company confirmed FDA’s guidance for a single-arm study and gained alignment with the agency on an alternative potential comparator study design for its monotherapy program. Based on feedback from the EMA and the FDA, the company now intend to conduct a single, randomized, controlled Phase 2 trial of elacestrant as a third-line monotherapy in approximately 300 patients with ER+/HER2- advanced/metastatic breast cancer. Patients in the study would be randomized to receive either elacestrant or the investigator’s choice of an approved hormonal agent and the primary endpoint of the study will be progression-free survival (“PFS”). The study would also include a planned interim PFS analysis. The company believe that, depending on results, this single trial would support applications for global marketing approvals for elacestrant as a third-line monotherapy. In addition, depending on results of the interim analysis, the Company could seek accelerated approval for elacestrant in the United States. The company will provide further study details when the Phase 2 study is started, which the company expect will be in the second half of 2018.
Phase 1 - Dose-Escalation and Expansion Study
In December 2014, the company commenced a Phase 1, multicenter, open-label, multiple-part, dose-escalation study of elacestrant in postmenopausal women with ER-positive and HER2-negative advanced breast cancer in the United States to determine the recommended dose for a Phase 2 clinical trial and to make a preliminary evaluation of the potential anti-tumor effect of elacestrant. Part A of this Phase 1 study was designed to evaluate escalating doses of elacestrant. The Part B expansion cohort was initiated at 400-mg daily dosing in March 2016 to allow for an evaluation of additional safety, tolerability and preliminary efficacy. The patients enrolled in this study are heavily pretreated ER-positive, HER2-negative advanced breast cancer patients who have received a median of 3 prior lines of therapy including fulvestrant and CDK4/6 inhibitors, and about 50% of the patients had ESR1 mutations. Radius Health has completed enrollment in the ongoing dose-escalation Part A and expansion study parts B and C. In December 2017, the company initiated enrollment of a Part D cohort in this study to provide additional data on a more homogeneous and genetically defined patient population to support its overall elacestrant clinical development program and anticipated regulatory submissions.
In December 2016 and June 2017, the company reported positive results from this ongoing Phase 1 dose-escalation and expansion study. As of the study cut-off date of April 28, 2017, the elacestrant single agent ORR, was 23% with five confirmed partial responses in heavily pre-treated patients with advanced ER-positive breast cancer and in the 400-mg patient group of 26 patients with mature data, the median progression free survival was 4.5 months. These results showed that elacestrant was well-tolerated with the most commonly reported adverse events being low grade nausea and dyspepsia. In December 2017, the company reported additional updated data from this ongoing Phase 1 dose-escalation and expansion study, which included mature data from 40 patients treated at the 400 mg dose in this study. As of the study cut-off date of October 30, 2017, the elacestrant single agent ORR, was 27.3% with six confirmed partial responses out of 22 patients with response evaluation criteria in solid tumors ("RECIST") measurable disease. The median progression free survival was 5.4 months and clinical benefit rate at 24 weeks was 47.4%. These results showed that elacestrant was well-tolerated with the most commonly reported adverse events being low grade nausea, dyspepsia and vomiting.
Phase 1 - FES-PET Study
In December 2015, the company commenced a Phase 1 FES-PET study in patients with metastatic breast cancer in the European Union, which includes the use of FES-PET imaging to assess estrogen receptor occupancy in tumor lesions following elacestrant treatment. In December 2016, the company reported positive results from the Phase 1 FES-PET study. The first three enrolled patients dosed at the 400-mg cohort had a tumor FES-PET signal intensity reduction ranging from 79% to 91% at day 14 compared to baseline. This study enrolled five additional patients in the 400-mg daily oral cohort, followed by eight patients in the 200-mg daily oral cohort.
In December 2017, the company reported updated data from the Phase 1 FES-PET study that elacestrant demonstrated robust reduction in tumor ER availability in patients with advanced ER+ breast cancer who progressed on prior endocrine therapy. Seven out of eight patients dosed at the 400-mg cohort, and four out of seven patients dosed at the 200-mg cohort, had a tumor FES-PET signal intensity reduction equal to, or greater than, 75% at day 14 compared to baseline. The reduction in FES uptake supports flexibility for both 200-mg and 400-mg elacestrant dose selection for further clinical development in combination studies with various targeted agents and was similar in patients harboring mutant or wild-type ESR1. The most commonly reported adverse events reported were grade 1 and 2 nausea and dyspepsia.
Potential for use in Combination Therapy
In July 2015, the company announced that early but promising preclinical data showed that its investigational drug elacestrant, in combination with Pfizer’s palbociclib, a cyclin-dependent kinase ("CDK 4/6 inhibitor") or Novartis’ everolimus, an mTOR inhibitor, was effective in shrinking tumors. In preclinical patient-derived xenograft breast cancer models with either wild type or mutant ESR1, treatment with elacestrant resulted in marked tumor growth inhibition, and the combination of elacestrant with either agent, palbociclib or everolimus, showed anti-tumor activity that was significantly greater than either agent alone. The company believe that this preclinical data suggests that elacestrant has the potential to overcome endocrine resistance, is well-tolerated, and has a profile that is well suited for use in combination therapy.
In December 2017, the company announced additional preclinical data that continues to demonstrate elacestrant anti-tumor activity, as a single agent and in combination, in multiple models. In these preclinical models, elacestrant demonstrated marked tumor growth inhibition, as a single agent in models treated with multiple rounds of fulvestrant and in combination with CDK 4/6 inhibitors such as palbociclib and abemaciclib and with a phosphoinositide 3-kinase inhibitor, alpelisib.
Collaborations
In July 2016, the company entered into a preclinical collaboration with Takeda Pharmaceutical Company Limited to evaluate the combination of elacestrant with Takeda's investigational drug TAK-228, an oral mTORC 1/2 inhibitor in Phase 2b development for the treatment of breast, endometrial and renal cancer, with the goal of potentially exploring such combination in a clinical study. The company and Takeda have each agreed to contribute resources and supply compound material necessary for studies to be conducted under the collaboration and will share third party out-of-pocket research and development expenses. Activities under this collaboration are ongoing. Upon completion, both parties will agree upon the appropriate communication of the results.
In January 2016, the company entered into a worldwide clinical collaboration with Novartis Pharmaceuticals to evaluate the safety and efficacy of combining elacestrant with Novartis’ investigational agent LEE011 (ribociclib), a CDK 4/6 inhibitor, and BYL719 (alpelisib), an investigational phosphoinositide 3-kinase inhibitor. In January 2018, the company terminated this collaboration following the completion of preclinical studies. Radius Health is evaluating additional opportunities to collaborate with companies to evaluate the safety and efficacy of combining elacestrant with other agents for the treatment of breast cancer. The company believe that such combinations may be suitable in earlier lines of treatment for patients with advanced disease.
Vasomotor Symptoms
In December 2017, following a strategic review, the company announced that the company decided to discontinue further evaluation of elacestrant for vasomotor symptoms to focus instead on the continued clinical development of the compound as a potential treatment option in breast cancer.
RAD140
RAD140 is an internally discovered SARM. The androgen receptor ("AR") is highly expressed in many ER-positive, ER-negative, and triple-negative receptor breast cancers. Due to its receptor and tissue selectivity, potent activity, oral bioavailability, and long half-life, the company believe RAD140 could have clinical potential in the treatment of breast cancer. The company hold worldwide commercialization rights to RAD140.
In July 2016, the company reported that RAD140 in preclinical xenograft models of breast cancer demonstrated potent tumor growth inhibition when administered alone or in combinations with CDK4/6 inhibitors. It is estimated that approximately 70% of breast cancers express the androgen receptor. The company's data suggest that RAD140 activity at the androgen receptor leads to activation of AR signaling pathways including an AR-specific tumor suppressor and suppression of ER signaling. In April 2017, the company presented these RAD140 preclinical results at a major scientific congress.
In September 2017, the company initiated a Phase 1 study of RAD140 in patients with hormone receptor positive locally advanced or metastatic breast cancer. The clinical trial is designed to evaluate the safety and maximum tolerated dose of RAD140 in approximately 40 patients. Primary safety outcomes from the trial include rate of dose-limiting toxicities, adverse events related to treatment, and tolerability as measured by dose interruptions or adjustments. In addition, pharmacokinetics, pharmacodynamics and tumor response will also be evaluated. The company expect to provide an update on its RAD140 development program by the end of 2018.
Strategy
To achieve its goal of becoming a leading provider of innovative endocrine therapeutics in the areas of osteoporosis and oncology, the company plan to:
- Become a market leader for anabolic osteoporosis therapies. TYMLOS was approved by the FDA in April 2017, with U.S. commercial sales commencing in May 2017, and are focused on growing its market share in anabolic appropriate patients. Radius Health is conducting additional clinical research towards potential additional indications for TYMLOS, including a clinical trial in men with osteoporosis that the company expect to initiate in the first quarter of 2018, and which, if successful, will form the basis of a supplemental NDA seeking to expand the use of TYMLOS to treat men with osteoporosis at high risk for fracture. In the first half of 2018, the company plan to initiate a bone histomorphometry study, which would enroll approximately 25 postmenopausal women with osteoporosis to evaluate the early effects of TYMLOS on tissue-based bone remodeling and structural indices.
- Selectively pursue partnerships or collaborations to commercialize abaloparatide-SC outside the U.S. In July 2017, the company entered into a license and development agreement with Teijin for abaloparatide-SC in Japan. Under this agreement, the company received an upfront payment and may receive up to an aggregate of $40.0 million upon the achievement of certain regulatory and sales milestones, and a fixed low double-digit royalty based on net sales of abaloparatide-SC in Japan during the royalty term. In addition, Radius Health has an option to negotiate for a co-promotion agreement with Teijin for abaloparatide-SC in Japan. The company intend to enter into a collaboration for the commercialization of abaloparatide-SC outside of the United States and Japan.
- Expand abaloparatide's market potential through the continued development of abaloparatide-patch. Radius Health is developing its investigational abaloparatide-patch as a short-wear-time transdermal patch. In January 2018, the company met with the FDA and gained alignment with the agency on a single, pivotal BMD non-inferiority bridging study to support an NDA submission. The FDA agreed that, depending on the study results, a randomized, open label, active-controlled, non-inferiority Phase 3 study of up to 500 patients with postmenopausal osteoporosis at high risk of fracture would be sufficient to gain approval for abaloparatide-patch. The FDA confirmed that the primary endpoint will be change in lumbar spine BMD at 12 months and that the non-inferiority margin must preserve 75% of the active control (abaloparatide-SC) based on the lower bound of the 95% confidence interval. The company expect to initiate this pivotal study in mid-2019 and to complete it in 2020. In February 2018, the company entered into a scale-up and commercial supply agreement with 3M Company pursuant to which 3M has agreed to exclusively manufacture Phase 3 and global commercial supplies of abaloparatide-patch.
- Become a leader in the field of hormone-receptor driven cancers. Radius Health is developing its investigational product candidate elacestrant as a potential treatment for hormone-receptor positive breast cancer as a single agent and in combination with other therapies. Based on feedback from the EMA and the FDA, the company intend to conduct a single, randomized, controlled Phase 2 trial of elacestrant as a third-line monotherapy in approximately 300 patients with ER+/HER2- advanced/metastatic breast cancer. The company believe that, depending on results, this single trial would support applications for global marketing approvals for elacestrant as a third-line monotherapy. In addition, depending on results of the interim analysis, the Company could seek accelerated approval for elacestrant in the United States. The company will provide further study details when the Phase 2 study is started, which the company expect will be in the second half of 2018. Radius Health is exploring potential strategic collaborations to broaden development to potentially address earlier lines of treatment in combination with other anti-cancer agents. In addition, Radius Health is leveraging its experience with elacestrant to develop a next generation SERD for potential use in the treatment of hormone-drive cancers. Radius Health is advancing the development of RAD140 as a potential treatment for hormone-receptor positive breast cancer and in September 2017 the company initiated a Phase 1 study of RAD140 in patients with locally advanced or metastatic breast cancer.
- Continue to expand its product portfolio. The company plan to leverage its drug development expertise to discover and develop additional investigational product candidates focused on osteoporosis, oncology and endocrine diseases and conditions. The company may also consider opportunistically expanding its product portfolio within these areas through in-licensing, acquisitions or partnerships.
Opportunity
Osteoporosis
Osteoporosis is a disease characterized by low bone mass and structural deterioration of bone tissue, which leads to greater fragility and an increase in fracture risk. All bones become more fragile and susceptible to fracture as the disease progresses. People tend to be unaware that their bones are getting weaker, and a person with osteoporosis can fracture a bone from even a minor fall. The debilitating effects of osteoporosis have substantial costs. Loss of mobility, admission to nursing homes and dependence on caregivers are all common consequences of osteoporosis. The prevalence of osteoporosis is growing and, per the National Osteoporosis Foundation ("NOF"), is significantly under-recognized and under-treated in the population. While the aging of the population is a primary driver of an increase in cases, osteoporosis is also increasing from the use of drugs that induce bone loss, such as chronic use of glucocorticoids and aromatase inhibitors that are increasingly used for breast cancer and hormone therapies used for prostate cancer.
The NOF has estimated that 10 million people in the United States, composed of eight million women and two million men, already have osteoporosis, and another approximately 44 million have low bone mass placing them at increased risk for osteoporosis. In addition, the NOF has estimated that osteoporosis is responsible for more than two million fractures in the United States each year resulting in an estimated $19 billion in costs annually. The NOF expects that the number of fractures in the United States due to osteoporosis will rise to three million by 2025, resulting in an estimated $25.3 billion in costs each year. Worldwide, osteoporosis affects an estimated 200 million women according to the International Osteoporosis Foundation ("IOF") and causes more than 8.9 million fractures annually, which is equivalent to an osteoporotic fracture occurring approximately every three seconds.
The IOF has estimated that 1.6 million hip fractures occur worldwide each year, and by 2050 this number could reach between 4.5 million and 6.3 million. The IOF estimates that in Europe alone, the annual cost of osteoporotic fractures could surpass €76 billion by 2050. The IOF, in its 2013 Asia-Pacific audit, estimated that osteoporosis affects 10% of the population in Japan over age 40; composed of 9.8 million women and 3 million men. By 2025, it is expected that 25% of Japan’s population will be over 70 years old with an average life expectancy of 87 years, and this is predicted to increase to 32% of Japan’s population in 2050 with an average life expectancy of 92 years. In 2050, it is also expected that over half of the Japanese population will be over 50 years old. The expected increase in the age of its population presents Japan with a significant need to focus on the health of its elderly, including osteoporosis. In 2017, total sales of branded osteoporosis drugs approximated $7.3 billion, worldwide, of which more than $4.3 billion was attributable to injectable therapies. There are two main types of osteoporosis drugs currently available in the United States, anti-resorptive agents and anabolic agents. Anti-resorptive agents act to prevent further bone loss by inhibiting the breakdown of bone, whereas anabolic agents stimulate bone formation to build new bone.
The company believe there is a large unmet need in the market for osteoporosis treatment because existing therapies have been reported to have shortcomings in efficacy, tolerability and convenience. For example, one current standard of care, bisphosphonates, which are anti-resorptive agents, have been associated with infrequent but serious adverse events, such as osteonecrosis of the jaw and atypical fractures, especially of long bones. These side effects, although uncommon, reportedly have created increasing concern with physicians and patients. The company believe many physicians are seeking alternatives to bisphosphonates. Forteo/Forsteo® (teriparatide) marketed by Eli Lilly and Company ("Lilly") and Prolia® (denosumab) marketed by Amgen Inc. ("Amgen") are two alternatives to bisphosphonates that are approved for the treatment of osteoporosis. Prolia has also been associated with infrequent but serious adverse events, such as osteonecrosis of the jaw and atypical fractures. In 2017, Forteo/Forsteo had reported worldwide sales of approximately $1.8 billion, $1.0 billion in the United States and $0.8 billion outside of the United States, and Prolia had reported worldwide sales of approximately $2.0 billion, $1.3 billion in the United States and $0.7 billion outside of the United States. In 2016, IMS (now IQVIA) estimated that sales for osteoporosis medicines in Japan were 292 billion yen, or approximately $2.7 billion, of which aggregate sales for anabolic agents (Lilly’s Forteo and Asahi Kasei’s Teribone) comprised 77 billion yen, or $720 million.
The company believe there is a significant opportunity for TYMLOS (abaloparatide), an anabolic agent which is approved in the U.S. for the treatment of postmenopausal women with osteoporosis at high risk for fracture defined as history of osteoporotic fracture, multiple risk factors for fracture, or patients who have failed or are intolerant to other available osteoporosis therapy. With the potential addition of new guidelines, expanding research, increased diagnosis effort, greater awareness of the long-term risk associated with osteoporotic fracture, and new, more effective therapies the company believe osteoporosis treatment will expand and likewise its potential commercial opportunity. The company also believe that there is a significant opportunity for abaloparatide outside the U.S., particularly in Japan, where Radius Health has a license and development agreement with Teijin for abaloparatide-SC under which Radius Health is entitled to receive payments up to an aggregate of $40.0 million upon the achievement of certain regulatory and sales milestones, a fixed low double-digit royalty based on net sales of abaloparatide-SC in Japan during the royalty term, and have an option to negotiate for a co-promotion agreement with Teijin for abaloparatide-SC in Japan.
Abaloparatide
In April 2017, the FDA approved TYMLOS for the treatment of postmenopausal women with osteoporosis at high risk for fracture defined as history of osteoporotic fracture, multiple risk factors for fracture, or patients who have failed or are intolerant to other available osteoporosis therapy. Radius Health is developing two formulations of abaloparatide: abaloparatide-SC and abaloparatide-patch.
Abaloparatide-SC
TYMLOS was approved in the United States in April 2017 for the treatment of postmenopausal women with osteoporosis at high risk for fracture. The first commercial sales of TYMLOS in the United States occurred in May 2017. Radius Health is commercializing TYMLOS in the United States through its commercial organization. Radius Health has a distribution network of well-established distributors and specialty pharmacies for TYMLOS in the United States. Under its distribution model, both the distributors and specialty pharmacies take physical delivery of TYMLOS and the specialty pharmacies dispense TYMLOS directly to patients. The company hold worldwide commercialization rights to abaloparatide-SC, except for Japan, where Radius Health has an option to negotiate a co-promotion agreement with Teijin for abaloparatide-SC.
The combined 25-month fracture data from its Phase 3 clinical trial program for TYMLOS formed the basis of its regulatory submissions in the United States and Europe. In November 2015, the company submitted an MAA for abaloparatide-SC to the EMA, which was validated and is currently undergoing active regulatory assessment by the CHMP. In December 2017, the CHMP issued a third Day-180 List of Outstanding Issues. As part of its on-going risk-benefit assessment, the CHMP informed the Company that it intends to refer the MAA to a scientific advisory group for additional advice. The company expect that the CHMP may adopt an opinion regarding its MAA during the first half of 2018. The company intend to enter into a collaboration for the commercialization of abaloparatide-SC outside of the United States and Japan.
In May 2017, the company announced positive top-line results from the completed 24-month ACTIVExtend clinical trial of TYMLOS, which met all its primary and secondary endpoints. In ACTIVExtend, patients who had completed 18 months of TYMLOS (abaloparatide) injections or placebo in the ACTIVE Phase 3 trial were transitioned to receive 24 additional months of open-label alendronate. For the subset of ACTIVE trial patients (n=1139) that enrolled in the ACTIVExtend trial, the previous TYMLOS-treated patients had a significant 84% relative risk reduction (p<0.0001) in the incidence of new vertebral fractures compared with patients who received placebo followed by alendronate. They also demonstrated a 39% risk reduction in nonvertebral fractures (p=0.038), a 34% risk reduction clinical fractures (p=0.045) and a 50% risk reduction in major osteoporotic fractures (p=0.011) compared with patients who received placebo followed by alendronate. At the 43-month timepoint, for all patients (n=1645) that enrolled in the ACTIVE trial, TYMLOS-treated patients had a statistically significant risk reduction in new vertebral fractures (p<0.0001), nonvertebral fractures (p=0.038), clinical fractures (p=0.045), and major osteoporotic fractures (p<0.001), compared with patients who received placebo followed by alendronate. While not a pre-specified endpoint, there was also a statistically significant risk reduction in hip fractures (p=0.027) at the 43-month time point in the TYMLOS-treated patients, compared with patients who received placebo followed by alendronate. The adverse events reported during the alendronate treatment period were similar between the previous TYMLOS-treated patients and the previous placebo group. The incidences of cardiovascular adverse events including serious adverse events were similar between groups. There have been no cases of osteonecrosis of the jaw or atypical femoral fracture in the entire TYMLOS development program. The results from the completed ACTIVExtend trial were presented at a major scientific meeting in September 2017 and the company submitted a labeling supplement in connection with this data to the FDA in December 2017.
In July 2017, the company entered into a license and development agreement with Teijin for abaloparatide-SC in Japan. Pursuant to the agreement, the company may receive additional milestone payments upon the achievement of certain regulatory and sales milestones, and a fixed low double-digit royalty based on net sales of abaloparatide-SC in Japan during the royalty term. In addition, Radius Health has an option to negotiate for a co-promotion agreement with Teijin for abaloparatide-SC in Japan.
In late 2017, the company gained agreement with the FDA on the design of a clinical trial in men with osteoporosis which, if successful, will form the basis of a supplemental NDA seeking to expand the use of TYMLOS to treat men with osteoporosis at high risk for fracture. The study will be a randomized, double-blind, placebo-controlled trial that will enroll approximately 225 men with osteoporosis. The primary endpoint is change in lumbar spine BMD at 12 months compared with placebo. In previous clinical trials, TYMLOS has demonstrated increases in BMD in postmenopausal women. The study will include specialized high-resolution imaging of bone structure in a subset of the study participants. The company expect to initiate the trial in the first quarter of 2018.
In the first half of 2018, the company plan to initiate a bone histomorphometry study, which would enroll approximately 25 postmenopausal women with osteoporosis to evaluate the early effects of TYMLOS on tissue-based bone remodeling and structural indices.
Abaloparatide-patch
Radius Health is also developing abaloparatide-patch, based on 3M’s patented Microstructured Transdermal System technology, for potential use as a short wear-time transdermal patch. The company hold worldwide commercialization rights to the abaloparatide-patch technology and Radius Health is developing abaloparatide-patch toward future global regulatory submissions to build upon the potential success of TYMLOS. The company's development strategy for abaloparatide patch is to bridge to the established efficacy and safety of its approved abaloparatide-SC formulation.
The company commenced a human replicative clinical evaluation of the optimized abaloparatide-patch in December 2015, with the goal of achieving comparability to abaloparatide-SC. In September 2016, the company presented results from this evaluation of the first and second abaloparatide-patch prototypes, demonstrating that formulation technology can modify the pharmacokinetic profile of abaloparatide, including Tmax, half-life (“T1/2”), and area under the curve (“AUC”). In March 2018, the company announced that through further optimization the company had achieved comparability to the abaloparatide-SC profile with a third prototype (the “current abaloparatide-patch”). The current abaloparatide-patch optimized the drug-device combination through process improvements, a finalized formulation, selection of a dose (300 µg), and the introduction of a new clinical applicator. Together these changes, which were designed to improve the ease of use and patient experience, resulted in an increased half-life and AUC (915 pg.hr/ml for the current abaloparatide-patch, compared to 242 pg.hr/ml for the first patch prototype, 645 pg.hr/ml for the second patch prototype, and 936 pg.hr/ml for abaloparatide-SC).
In January 2018, the company met with the FDA to align on a regulatory and development path for registration of abaloparatide-patch. The company gained alignment with the agency on a single, pivotal BMD non-inferiority bridging study to support an NDA submission. The FDA agreed that, depending on the study results, a randomized, open label, active-controlled, non-inferiority Phase 3 study of up to 500 patients with postmenopausal osteoporosis at high risk of fracture would be sufficient to gain approval for abaloparatide-patch. The FDA confirmed that the primary endpoint will be change in lumbar spine BMD at 12 months and that the non-inferiority margin must preserve 75% of the active control (abaloparatide-SC) based on the lower bound of the 95% confidence interval. The company expect to initiate this pivotal study in mid-2019. On February 27, 2018, the company entered into a scale-up and commercial supply agreement with 3M Company pursuant to which 3M has agreed to exclusively manufacture Phase 3 and global commercial supplies of abaloparatide-patch.
Breast Cancer
According to the World Health Organization, breast cancer is the most common cancer in women and the global incidence is expected to increase in the coming years. The major cause of death from breast cancer is metastases, most commonly to the bone, liver, lung and brain. Approximately 30% of early-stage breast cancer patients develop metastatic disease, and of those patients 90% relapse during the course of their treatment. About 5% of breast cancer patients have distant metastases at the time of diagnosis. Patients with metastatic breast cancer have a five-year survival rate of only 25%, compared with a greater than 90% five-year survival rate for patients with only local disease at diagnosis. Importantly, even patients without metastases at diagnosis are at risk for developing metastases over time.
Approximately 70% of breast cancers express the ER and depend on estrogen signaling for growth and survival. The standard of care for ER+ advanced/metastatic breast cancer calls for endocrine therapy at all stages of treatment, with patients typically cycling through multiple anti-estrogen therapies, such as aromatase inhibitors (“AIs”), selective estrogen receptor modulators (“SERMs”), and selective estrogen receptor degraders (“SERDs”).
These therapies inhibit the ER pathway either by inhibiting estrogen synthesis (AIs) or by directly inhibiting the estrogen receptor (SERMs and SERDs). While both SERMs and SERDs antagonize the estrogen receptor, SERDs function to degrade the receptor. Although many patients initially respond to AIs and SERMs, a majority of patients will have progressive disease and the dependence on ER for tumor growth and sensitivity to other ER-targeting agents is often retained. On the basis of this continued dependence on ER, novel SERDs have gained widespread attention as a means of delivering more durable responses and increasing progression-free survival in this setting. Indeed, SERDs have demonstrated clinical efficacy in patients who have progressed on AIs or SERMs.
Currently only one SERD, fulvestrant, an intramuscular injection, is approved for the treatment of ER-positive metastatic breast cancer. The company believe a significant opportunity may exist for new oral therapies that can more effectively treat ER-positive breast cancer.
Elacestrant (RAD1901)
Elacestrant (RAD1901) is a SERD that Radius Health is evaluating for potential use as a once daily oral treatment for hormone-receptor positive breast cancer. The company hold worldwide commercialization rights to elacestrant, which the company licensed from Eisai Co. Ltd. ("Eisai"). Elacestrant is currently being investigated in women with ER-positive and HER2-negative breast cancer, the most common subtype of the disease. Studies completed to date indicate that the compound has the potential for use as a single agent or in combination with other therapies for the treatment of breast cancer. To date, no dose limiting toxicities have been reported in the elacestrant program.
The company believe that, subject to successful development, regulatory review and approval, elacestrant could have the potential to offer the following advantages over other current standard of care treatments for ER-positive breast cancer:
- ability to degrade the estrogen receptor;
- avorable efficacy and tolerability profile;
- ability to effectively combine with other agents;
- treatment of hormone-resistant breast cancers; and
- once a day oral administration.
Radius Health has completed enrollment in its 18-F fluoroestradiol positron emission tomography (“FES-PET”) imaging study and dose-escalation Part A and expansion study parts B and C Phase 1 breast cancer trials. In June 2017, the company discussed the data from these ongoing Phase 1 studies with the FDA to gain alignment on defining the next steps for its elacestrant breast cancer program, including the design of a Phase 2 trial. In this meeting, the FDA agreed that a single-arm monotherapy Phase 2 study of up to 200 patients, could be appropriate with the primary endpoint being ORR, coupled with DOR. Depending on the study results, which must demonstrate an improvement over then available therapies, this study could be considered a pivotal study for accelerated approval as long as a confirmatory study is ongoing at the time of its NDA submission. In October 2017, the FDA granted Fast Track designation for its elacestrant breast cancer program.
In February 2018, the company received scientific advice from the European Medicines Agency (“EMA”) regarding a potential single-arm monotherapy Phase 2 trial of elacestrant in patients with ER+, HER2- advanced or metastatic breast cancer. In addition, the company had a further meeting in February 2018 with the FDA regarding the registrational pathway for elacestrant at which the company confirmed FDA’s guidance for a single-arm study and gained alignment with the agency on an alternative potential comparator study design for its monotherapy program. Based on feedback from the EMA and the FDA, the company now intend to conduct a single, randomized, controlled Phase 2 trial of elacestrant as a third-line monotherapy in approximately 300 patients with ER+/HER2- advanced/metastatic breast cancer. Patients in the study would be randomized to receive either elacestrant or the investigator’s choice of an approved hormonal agent and the primary endpoint of the study will be progression-free survival (“PFS”). The study would also include a planned interim PFS analysis. The company believe that, depending on results, this single trial would support applications for global marketing approvals for elacestrant as a third-line monotherapy. In addition, depending on results of the interim analysis, the Company could seek accelerated approval for elacestrant in the United States. The company will provide further study details when the Phase 2 study is started, which the company expect will be in the second half of 2018.
Phase 1 - Dose-Escalation and Expansion Study
In December 2014, the company commenced a Phase 1, multicenter, open-label, multiple-part, dose-escalation study of elacestrant in postmenopausal women with ER-positive and HER2-negative advanced breast cancer in the United States to determine the recommended dose for a Phase 2 clinical trial and to make a preliminary evaluation of the potential anti-tumor effect of elacestrant. Part A of this Phase 1 study was designed to evaluate escalating doses of elacestrant. The Part B expansion cohort was initiated at 400-mg daily dosing in March 2016 to allow for an evaluation of additional safety, tolerability and preliminary efficacy. The patients enrolled in this study are heavily pretreated ER-positive, HER2-negative advanced breast cancer patients who have received a median of 3 prior lines of therapy including fulvestrant and CDK4/6 inhibitors, and about 50% of the patients had ESR1 mutations. Radius Health has completed enrollment in the ongoing dose-escalation Part A and expansion study parts B and C. In December 2017, the company opened a Part D cohort in this study to provide additional data on a more homogeneous and genetically defined patient population to support its overall elacestrant clinical development program and anticipated regulatory submissions.
In December 2016, the company reported positive results from this ongoing Phase 1 dose-escalation and expansion study. These results showed that elacestrant was well-tolerated with the most commonly reported adverse events being low grade nausea and dyspepsia. Enrollment in the Part C tablet dosage form cohort was completed in November 2016.
In June 2017, the company reported additional positive data from this ongoing Phase 1 dose-escalation and expansion study. As of the study cut-off date of April 28, 2017, the elacestrant single agent ORR, was 23% with five confirmed partial responses in heavily pre-treated patients with advanced ER-positive breast cancer. In the 400-mg patient group of 26 patients with mature data, the median progression free survival was 4.5 months. These results showed that elacestrant was well-tolerated with the most commonly reported adverse events being low grade nausea and dyspepsia.
In December 2017, the company reported updated data from this ongoing Phase 1 dose-escalation and expansion study, which included mature data from 40 patients treated at the 400 mg dose in this study. As of the study cut-off date of October 30, 2017, the elacestrant single agent ORR, was 27.3% with six confirmed partial responses out of 22 patients with RECIST measurable disease. The median progression free survival was 5.4 months and clinical benefit rate at 24 weeks was 47.4%. These results showed that elacestrant was well-tolerated with the most commonly reported adverse events being low grade nausea, dyspepsia and vomiting.
Phase 1 - FES-PET Study
In December 2015, the company commenced a Phase 1 18-F fluoroestradiol positron emission tomography ("FES-PET") study in patients with metastatic breast cancer in the European Union, which includes the use of FES-PET imaging to assess estrogen receptor occupancy in tumor lesions following elacestrant treatment. In December 2016, the company reported positive results from the Phase 1 FES-PET study. The first three enrolled patients dosed at the 400-mg cohort had a tumor FES-PET signal intensity reduction ranging from 79% to 91% at day 14 compared to baseline. This study enrolled 5 additional patients in the 400-mg daily oral cohort, followed by 8 patients in the 200-mg daily oral cohort.
In December 2017, the company reported updated data from the Phase 1 FES-PET study that elacestrant demonstrated robust reduction in tumor ER availability in patients with advanced ER+ breast cancer who progressed on prior endocrine therapy. Seven out of eight patients dosed at the 400-mg cohort, and four out of seven patients dosed at the 200-mg cohort, had a tumor FES-PET signal intensity reduction equal to, or greater than, 75% at day 14 compared to baseline. The reduction in FES uptake supports flexibility for both 200-mg and 400-mg elacestrant dose selection for further clinical development in combination studies with various targeted agents and was similar in patients harboring mutant or wild-type ESR1. The most commonly reported adverse events reported were grade 1 and 2 nausea and dyspepsia.
Potential for use in Combination Therapy
In July 2015, the company announced that early but promising preclinical data showed that its investigational drug elacestrant, in combination with Pfizer’s palbociclib, a cyclin-dependent kinase 4/6 inhibitor, or Novartis’ everolimus, an mTOR inhibitor, was effective in shrinking tumors. In preclinical patient-derived xenograft breast cancer models with either wild type or mutant ESR1, treatment with elacestrant resulted in marked tumor growth inhibition, and the combination of elacestrant with either agent, palbociclib or everolimus, showed anti-tumor activity that was significantly greater than either agent alone. The company believe that this preclinical data suggests that elacestrant has the potential to overcome endocrine resistance, is well-tolerated, and has a profile that is well suited for use in combination therapy.
In December 2017, the company announced additional preclinical data that continues to demonstrate elacestrant anti-tumor activity, as a single agent and in combination, in multiple models. In these preclinical models, elacestrant demonstrated marked tumor growth inhibition, as a single agent in models treated with multiple rounds of fulvestrant and in combination with CDK 4/6 inhibitors such as palbociclib and abemaciclib and with a phosphoinositide 3-kinase inhibitor, alpelisib.
Collaborations
In July 2016, the company entered into a preclinical collaboration with Takeda Pharmaceutical Company Limited to evaluate the combination of elacestrant with Takeda's investigational drug TAK-228, an oral mTORC 1/2 inhibitor in Phase 2b development for the treatment of breast, endometrial and renal cancer, with the goal of potentially exploring such combination in a clinical study. The company and Takeda have each agreed to contribute resources and supply compound material necessary for studies to be conducted under the collaboration and will share third party out-of-pocket research and development expenses. Activities under this collaboration are ongoing. Upon completion, both parties will agree upon the appropriate communication of the results.
In January 2016, the company entered into a worldwide clinical collaboration with Novartis Pharmaceuticals to evaluate the safety and efficacy of combining elacestrant with Novartis’ investigational agent LEE011 (ribociclib), a CDK 4/6 inhibitor, and BYL719 (alpelisib), an investigational phosphoinositide 3-kinase inhibitor. In January 2018, the company terminated this collaboration following the completion of pre-clinical studies. Radius Health is evaluating additional opportunities to collaborate with companies to evaluate the safety and efficacy of combining elacestrant with other agents for the treatment of breast cancer. The company believe that such combinations may be suitable in earlier lines of treatment for patients with advanced disease.
Investigational Drug—RAD140
RAD140 is an internally discovered SARM. The androgen receptor is highly expressed in many ER-positive, ER-negative, and triple-negative receptor breast cancers. Due to its receptor and tissue selectivity, potent activity, oral bioavailability, and long half-life, the company believe RAD140 could have clinical potential in the treatment of breast cancer. The company hold worldwide commercialization rights to RAD140.
In September 2017, the company initiated a Phase 1 study of RAD140 in patients with hormone receptor positive locally advanced or metastatic breast cancer. The clinical trial is designed to evaluate the safety and maximum tolerated dose of RAD140 in approximately 40 patients. Primary safety outcomes from the trial include rate of dose-limiting toxicities, adverse events related to treatment, and tolerability as measured by dose interruptions or adjustments. In addition, pharmacokinetics, pharmacodynamics and tumor response will also be evaluated. The company expect to provide an update on its RAD140 development program by the end of 2018.
In July 2016, the company reported that RAD140 in preclinical xenograft models of breast cancer demonstrated potent tumor growth inhibition when administered alone or in combinations with CDK4/6 inhibitors. It is estimated that approximately 70% of breast cancers express the androgen receptor. The company's data suggest that RAD140 activity at the androgen receptor leads to activation of AR signaling pathways including an AR-specific tumor suppressor and suppression of ER signaling. In April 2017, the company presented these RAD140 preclinical results at a major scientific congress.
Manufacturing
The company do not own or operate manufacturing facilities for the production of its commercial product, TYMLOS, or any of its investigational product candidates, nor do Radius Health has plans to develop its own manufacturing operations in the foreseeable future.
Abaloparatide, the active pharmaceutical ingredient (“API”) for both TYMLOS and abaloparatide-patch, is manufactured for it on a contract basis by Polypeptide Laboratories Holding (PPL) AB (“PPL”), as successor-in-interest to Lonza Group Ltd., using a solid phase peptide synthesis assembly process, and purification by high pressure liquid chromatography. Abaloparatide for TYMLOS is supplied as a liquid in a multi-dose cartridge for use in a pen delivery device. The components of the pen delivery device are manufactured by Ypsomed AG (“Ypsomed”). The multi-dose cartridges and pen delivery device are filled, assembled and packaged by Vetter International GmbH (“Vetter”).
Abaloparatide-patch drug product is manufactured by 3M Company and 3M Innovative Properties Company, (together “3M”), based on their patented microneedle technology to administer drugs through the skin, as an alternative to subcutaneous injection.
Elacestrant API and drug product are manufactured for it on a contract basis by Patheon, Inc.
RAD140 API and drug product are manufactured for it on a contract basis by Alcami Corporation.
Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, which govern the methods used in, and the facilities and controls used for, the manufacture, processing, packing and holding of drugs. FDA and International Conference on Harmonisation ("ICH") current Good Manufacturing Practice (“cGMP”) requirements include those pertaining to record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. The company's contract manufacturing organizations are required to manufacture TYMLOS and its investigational product candidates under cGMP conditions. cGMP is a regulatory standard for the production of human pharmaceuticals that imposes extensive substantive, procedural and record keeping requirements on the manufacturing processes, testing methodology, and associated production and testing facilities.
Intellectual Property
As of December 31, 2017, the company owned or co-owned 11 issued U.S. patents, as well as 15 pending U.S. patent applications, 4 pending Patent Cooperation Treaty ("PCT") applications, 55 pending foreign patent applications in the European Patent Office and 14 other jurisdictions, and 99 granted foreign patents. As of December 31, 2017, the company had licenses to 3 U.S. patents related to compositions and related uses thereof, as well as numerous foreign counterparts to many of these patents and patent applications. The company own the federal trademark registration in the United States for Radius® in association with pharmaceuticals. In addition, Radius Health has received notices of allowance in the U.S. and in Canada for TYMLOS and for trademarks on potential brand names for its product candidates in the U.S. and in other countries.
The company strive to protect the proprietary technology that the company believe is important to its business, including seeking and maintaining patents intended to cover its investigational product candidates and compositions, their methods of use and processes for their manufacture and any other inventions that are commercially important to the development of its business. The company also rely on trade secrets to protect aspects of its business that are not amenable to, or that the company do not consider appropriate for, patent protection.
The company's success will significantly depend on its ability to obtain and maintain patent and other proprietary protection for commercially important technology and inventions and know-how related to its business, defend and enforce its patents, preserve the confidentiality of its trade secrets, and operate without infringing the valid and enforceable patents and proprietary rights of third parties. The company also rely on know-how and continuing technological innovation to develop and maintain its proprietary position.
Abaloparatide
The company acquired and maintain exclusive worldwide rights, excluding development and commercialization rights for Japan, to certain patents, data and technical information related to abaloparatide through a license agreement with an affiliate of Ipsen Pharma SAS ("Ipsen"). Composition of matter of abaloparatide was claimed in the United States (U.S. Patent No. 5,969,095), Europe, Australia, Canada, China, Hong Kong, South Korea, New Zealand, Poland, Russia, Singapore, Mexico, Hungary, and Taiwan. These patents and European Patent No. 0847278, which was included in the license from Ipsen and claimed the composition of matter of abaloparatide, expired in 2016. The subcutaneous formulation of abaloparatide for use in treating osteoporosis is covered by Patent No. 7,803,770 until the statutory term expires October 3, 2027, which the company expect will be extended to March 26, 2028 (statutory term that may be extended with 175 days of patent term adjustment due to delays in patent prosecution by the United States Patent and Trademark Office, or USPTO) in the United States (not including any patent term extension under the Hatch-Waxman Act). The intended therapeutic formulation for abaloparatide-SC is covered by Patent No. 8,148,333 until 2027 in the United States (not including any patent term extension under the Hatch-Waxman Act). Related patents granted in Europe, Australia, China, Israel, Japan, South Korea, Mexico, New Zealand, Russia, Singapore, and Ukraine, and additional patent applications pending in Brazil, Canada, Europe, Hong Kong, India, South Korea, and Norway, will have a patent expiration date of 2027, not taking into account extension under any applicable laws. A notice to grant the intended therapeutic formulation for abaloparatide-SC and its use for osteoporosis treatment has been received from the European Patent Office in February 2018. When granted, this patent and the granted European Patent No. 2957278 will have a normal expiry of October 3, 2027, not including any issued supplementary patent certificates ("SPC"). Radius Health is aware of two oppositions to European Patent No. 2957278 filed before the opposition period expired on February 19, 2018. One opposition was filed on February 16, 2018 by Teva Pharmaceutical Industries Ltd and the other opposition was filed on February 19, 2018 in the name of a patent law firm, Isenbruck Bosl Horschler LLP. Patent applications covering various aspects of abaloparatide for microneedle application have been granted in Australia, Europe, Japan, and New Zealand, and additional patent applications are currently pending in the United States, Europe, Hong Kong, and Japan. The issued patents and any patents that might issue from the pending applications will have statutory expiration dates ranging from 2032 to 2037, not taking into account extension under any applicable laws. Radius Health has worldwide rights to commercialize abaloparatide-patch, including in Japan.
Elacestrant (RAD1901)
The company exclusively licensed the worldwide rights to elacestrant from Eisai. U.S. Patent No. 7,612,114 (statutory term expires December 25, 2023 which may be extended up to August 18, 2026 with 967 days of patent term adjustment not taking into account any Hatch-Waxman patent term extensions) that covers elacestrant as a composition of matter as well as the use of elacestrant for treatment of estrogen-dependent breast cancer. Corresponding patents issued in Australia, Canada, Japan, Poland, and Europe and pending in India will have a statutory expiration date in 2023, not taking into account extension under any applicable laws. The company exclusively licensed US 9,421,264 (statutory term expires October 10, 2034) covering the treatment of ER+, SERM-resistant (such as tamoxifen and fulvestrant) breast cancer brain metastasis with elacestrant and related applications covering, more broadly, the use of elacestrant for the treatment of ER+ cancers, such as SERM-resistant ER+ breast cancer (statutory term expires October 10, 2034). Corresponding applications pending in Europe and Canada will have a statutory expiration date in 2035. Polymorphic forms of elacestrant are covered in a U.S. application and a PCT application (filed January 2018) having a projected statutory expiration date in 2038, not taking into account any extension under any applicable laws. Elacestrant combination therapies with a CDK4/6 inhibitor (e.g., palbociclib) or an mTOR inhibitor (e.g., everolimus) for treatment of cancers that are drug-resistant and/or expressing mutant ERα+ are covered by applications pending in the U.S., Australia, Brazil, Canada, China, Europe, Israel, Japan, South Korea, Mexico, New Zealand, Russia, and Singapore (statutory expiration date in 2036, not taking into account any extension under any applicable laws). RAD140
The composition of matter of, and methods of using, RAD140 are covered by U.S. Patent No. 8,067,448 (statutory term expires February 19, 2029, which the company expect will be extended to September 25, 2029, with potentially 218 days of patent term adjustment due to delays by the USPTO, not taking into account any Hatch Waxman patent term extensions) and U.S. Patent No. 8,268,872 (statutory term expires February 19, 2029 which may be extended to September 25, 2029 with patent term adjustment, subject to a terminal disclaimer of Patent Nos. 8,067,448 and 8,455,525). Related patents have been granted in Australia, Canada, Europe, Japan and Mexico and additional patent applications are pending in Brazil and India. Any patents issued from these filings will have a statutory expiration date in 2029. RAD140 for the treatment of breast cancer expressing the androgen receptor (“AR+ breast cancer”) is covered in a PCT application (projected statutory expiration date in 2037, not taking into account extension under any applicable laws). The PCT application covers the use of RAD140 alone or in combination with a CDK4/6 inhibitor (e.g., palbociclib) or an mTOR inhibitor (e.g., everolimus) for the treatment of the AR+ breast cancer.
There can be no assurance that an issued patent will remain valid and enforceable in a court of law through the entire patent term. Should the validity of a patent be challenged, the legal process associated with defending the patent can be costly and time consuming. Issued patents can be subject to oppositions, interferences and other third-party challenges that can result in the revocation of the patent or that can limit patent claims such that patent coverage lacks sufficient breadth to protect subject matter that is commercially relevant. Competitors may be able to circumvent its patents. Development and commercialization of pharmaceutical products can be subject to substantial delays and it is possible that at the time of commercialization any patent covering the product has expired or will be in force for only a short period of time following commercialization. The company cannot predict with any certainty if any third-party U.S. or foreign patent rights, or other proprietary rights, will be deemed infringed by the use of its technology. Nor can the company predict with certainty which, if any, of these rights will or may be asserted against it by third-parties. Should the company need to defend itself and its partners against any such claims, substantial costs may be incurred. Furthermore, parties making such claims may be able to obtain injunctive or other equitable relief, which could effectively block its ability to develop or commercialize some or all its products in the United States and abroad and could result in the award of substantial damages. In the event of a claim of infringement, the company or its partners may be required to obtain one or more licenses from a third party. There can be no assurance that the company can obtain a license on a reasonable basis should the company deem it necessary to obtain rights to an alternative technology that meets its needs. The failure to obtain a license may have a material adverse effect on its business, results of operations and financial condition.
The company also rely on trade secret protection for its confidential and proprietary information. No assurance can be given that the company can meaningfully protect its trade secrets on a continuing basis. Others may independently develop substantially equivalent confidential and proprietary information or otherwise gain access to its trade secrets.
It is its policy to require its employees and consultants, outside scientific collaborators, sponsored researchers and other advisors who receive confidential information from it to execute confidentiality agreements upon the commencement of employment or consulting relationships. These agreements provide that all confidential information developed or made known to these individuals during the course of the individual's relationship with it is to be kept confidential and is not to be disclosed to third parties except in specific circumstances. The agreements provide that all inventions conceived by an employee shall be its property. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for its trade secrets in the event of unauthorized use or disclosure of such information.
The company's success will depend in part on its ability to obtain and maintain patent protection, preserve trade secrets, prevent third parties from infringing upon its proprietary rights and operate without infringing upon the proprietary rights of others, both in the United States and other territories worldwide.
Competition
The development and commercialization of new products to treat the targeted indications of its investigational product candidates is highly competitive, and TYMLOS faces, and its product candidates if approved, will face considerable competition from major pharmaceutical, biotechnology and specialty pharmaceutical companies, including Lilly, Amgen, UCB S.A., Merck & Co, Novartis, Pfizer, Roche, Asahi Kasei, and Corium, that currently market and/or are seeking to develop products for similar indications. Many of its competitors have substantially more resources than the company do, including financial, manufacturing, marketing, research and drug development resources. In addition, many of these companies have longer operating histories and more experience than it in preclinical and clinical development, manufacturing, regulatory and global commercialization.
Abaloparatide
There are two main types of osteoporosis drugs currently available in the United States, anti-resorptive agents and anabolic agents. Anti-resorptive agents including bisphosphonates, estrogen, SERMs and Amgen's Prolia are the most common treatments for osteoporosis. Teriparatide, marketed by Lilly under the name Forteo/Forsteo (outside the U.S.), is the only other anabolic drug approved in the United States and the only anabolic drug approved Europe for the treatment of osteoporosis. Radius Health is aware of companies pursuing development of biosimilar and/or generic versions of teriparatide through various regulatory pathways, including Pfenex, Inc. in the United States; Teva Pharmaceutical Industries, Ltd., approved in various European Union member states and under U.S. regulatory review; and STADA Arzneimittel AG and Gedeon Richter, approved in the European Union (in each case, with launch not expected until expiration of the applicable patents covering teriparatide, or if earlier, invalidation of such patents in connection with currently pending patent litigation and/or challenges). Other companies are in earlier stages of development of a generic version of teriparatide. Other organizations are also working to develop new therapies to treat osteoporosis. For example, Amgen and UCB are co-developing romosozumab, a humanized monoclonal antibody that inhibits the action of sclerostin, for which an MAA has recently been filed and validated in the European Union.
In addition, Radius Health is aware that Corium is developing a transdermal form of PTH (1-34) that would compete with abaloparatide-patch.
Elacestrant (RAD1901)
Elacestrant for the treatment of hormone receptor positive breast cancer will face competition from SERDs, CNS-penetrant anti-cancer agents and from chemotherapy derivatives. AstraZeneca's Faslodex is the only SERD currently approved in the United States for the treatment of metastatic breast cancer. In addition, there are other organizations working to develop new therapies to treat metastatic breast cancer, including Roche, which is developing two oral SERD's which are currently in Phase 1 and Phase 2 clinical development.
RAD140
RAD140 is being developed for women with hormone receptor positive breast cancer. While no SARMs are currently approved as therapeutics in the United States, there are select competitive molecules in development across a range of indications, including in oncology (GTx), hip fractures (Viking Therapeutics), and cachexia (GSK).
The company cannot assure you that any of its current investigational product candidates, if successfully developed and approved, will be able to compete effectively against these, or any other competing therapeutics that may become available on the market.
Collaborations and License Agreements
3M
On February 27, 2018 the company entered into a Scale-Up And Commercial Supply Agreement (the “Supply Agreement”) with 3M, pursuant to which 3M has agreed to exclusively manufacture Phase 3 and global commercial supplies of an abaloparatide-coated transdermal patch product (“Product”) and associated applicator devices (“Applicator”). Under the Supply Agreement, 3M will manufacture Product and Applicator for it according to agreed-upon specifications in sufficient quantities to meet its projected supply requirements. 3M will manufacture commercial supplies of Product at unit prices that decrease with an increase in the quantity the company order. The company will pay 3M a mid-to-low single digit royalty on worldwide net sales of Product and reimburse 3M for certain capital expenditures incurred to establish commercial supply of Product. Radius Health is responsible for providing, at its expense, supplies of abaloparatide drug substance to be used in manufacturing Product. During the term of the Supply Agreement, 3M and Radius have agreed to work exclusively with each other with respect to the delivery of abaloparatide, parathyroid hormone (“PTH”), and/or PTH related proteins via active transdermal, intradermal, or microneedle technology.
The initial term of the Supply Agreement began on its effective date and will continue for five years after the first commercial sale of Product. The Supply Agreement then automatically renews for successive three-year terms, unless earlier terminated pursuant to its terms or upon either party’s notice of termination to the other 24 months prior to the end of the then-current term. The Supply Agreement may be terminated by either party upon an uncured material breach of its terms by the other party, or due to the other party’s bankruptcy, insolvency, or dissolution. Radius may terminate the Supply Agreement upon the occurrence of certain events, including for certain clinical, technical, or commercial reasons impacting Product, if Radius Health is unable to obtain U.S. regulatory approval for Product within a certain time period, or if the company cease development or commercialization of Product. 3M may terminate the Supply Agreement upon the occurrence of certain events, including if there are certain safety issues related to Product, if Radius Health is unable to obtain U.S. regulatory approval for Product within a certain time period, or if the company fail to order Product for a certain period of time after commercial launch of the Product in the U.S. Upon certain events of termination, 3M is required to transfer the manufacturing processes for Product and Applicator to it or a mutually agreeable third party and continue supplying Product and Applicator for a period of time pursuant to its projected supply requirements.
In June 2009, the company entered into a Development and Clinical Supplies Agreement with 3M, as amended (the “Development Agreement”), under which Product and Applicator development activities occur and 3M has manufactured phase 1 and 2 clinical trial supplies for it on an exclusive basis. The term of the Development Agreement runs until June 2019 and then automatically renews for additional one-year terms, unless earlier terminated, until the earliest of ![]() the expiration or termination of the Supply Agreement, (ii) the mutual written agreement of the parties, or (iii) prior written notice by either party to the other party at least ninety days prior to the end of the then-current term of the Development Agreement that such party declines to extend the term. Either party may terminate the agreement in the event of an uncured material breach by the other party. The company pay 3M for services delivered pursuant to the agreement on a fee-for-service or a fee-for-deliverable basis as specified in the agreement. Radius Health has paid 3M approximately $20.9 million, in the aggregate, through December 31, 2017 with respect to services and deliverables delivered pursuant to the 3M Agreement.
the expiration or termination of the Supply Agreement, (ii) the mutual written agreement of the parties, or (iii) prior written notice by either party to the other party at least ninety days prior to the end of the then-current term of the Development Agreement that such party declines to extend the term. Either party may terminate the agreement in the event of an uncured material breach by the other party. The company pay 3M for services delivered pursuant to the agreement on a fee-for-service or a fee-for-deliverable basis as specified in the agreement. Radius Health has paid 3M approximately $20.9 million, in the aggregate, through December 31, 2017 with respect to services and deliverables delivered pursuant to the 3M Agreement.
Ipsen Pharma
In September 2005, the company entered into a license agreement with Ipsen, as amended (the "License Agreement") under which the company exclusively licensed certain Ipsen compound technology and related patents covering abaloparatide to research, develop, manufacture and commercialize certain compounds and related products in all countries, except Japan (where Radius Health has an option to negotiate a co-promotion agreement for abaloparatide-SC) and France (where its commercialization rights were subject to certain co-marketing and co-promotion rights exercisable by Ipsen, provided that certain conditions included in the License Agreement were met). The company believe that Ipsen's co-marketing and co-promotion rights in France have permanently expired. Ipsen also granted it an exclusive right and license under the Ipsen compound technology and related patents to make and have made compounds or product in Japan. Ipsen further granted it an exclusive right and license under certain Ipsen formulation technology and related patents solely for purposes of enabling it to develop, manufacture and commercialize compounds and products covered by the compound technology license in all countries, except Japan and France (as discussed above).
In consideration for the rights to abaloparatide, and in recognition of certain milestones having been met to date, Radius Health has paid to Ipsen an aggregate amount of $13.0 million. The license agreement further requires it to make payments upon the achievement of certain future clinical and regulatory milestones. Total additional milestone payments that could be payable under the agreement as of December 31, 2017 are €24.0 million (approximately $28.7 million). The agreement provides that the company or its sublicensees are obligated to pay to Ipsen a fixed five percent royalty based on net sales of products containing abaloparatide on a country-by-country basis until the later of the last to expire of the licensed patents or for a period of 10 years after the first commercial sale of the licensed products in such country. The date of the last to expire of the abaloparatide patents licensed from or co-owned with Ipsen, barring any extension thereof, is expected to be March 26, 2028. In the event that the company sublicense abaloparatide to a third party, the agreement provides that the company would pay a percentage of certain payments received from such sublicensee (in lieu of milestone payments not achieved at the time of such sublicense). The applicable percentage is in the low double digit range. In addition, if the company or its sublicensees commercialize a product that includes a compound discovered by it based on or derived from confidential Ipsen know-how, the agreement provides that the company would pay to Ipsen a fixed low single digit royalty on net sales of such product on a country-by-country basis until the later of the last to expire of its patents that cover such product or for a period of 10 years after the first commercial sale of such product in such country.
The License Agreement expires on a country-by-country basis on the later of (1) the date the last remaining valid claim in the licensed patents expires in that country, or (2) a period of 10 years after the first commercial sale of the licensed products in such country, unless it is sooner terminated in accordance with its terms.
The License Agreement may be terminated by it with prior notice to Ipsen. The License Agreement may be terminated by Ipsen upon notice to it with immediate effect, if we, in any country of the world, bring an action or proceeding to challenge any Ipsen patent. The License Agreement can also be terminated by Ipsen if the company fail to use reasonable commercial efforts to develop the licensed product for sale and commercialization in those countries within the territory where it is commercially reasonable to do so as contemplated by the License Agreement, or fail to use reasonable commercial efforts to perform its obligations under the latest revised version of the development plan approved by the joint steering committee, or fail to use reasonable commercial efforts to launch and sell one licensed product in those countries within the territory where it is commercially reasonable to do so. Either party may also terminate the License Agreement upon an uncured material breach by the other party. Ipsen may terminate the License Agreement if the License Agreement is assigned or sublicensed, if a third party acquires it, or if the company acquire control over a PTH or a PTHrP compound that is in clinical development or is commercially available in the territory, and if following such assignment, sublicense, acquisition, or acquisition of control by it, such assignee, sublicensee, acquirer or we, fail to meet the timetable under the latest revised version of the development plan approved by the joint steering committee under the License Agreement.
Prior to executing the License Agreement for abaloparatide with Radius, Ipsen licensed the Japanese rights for abaloparatide to Teijin Limited ("Teijin") a Japanese pharmaceutical company. Teijin has initiated a Phase 3 clinical study of abaloparatide in Japan for the treatment of postmenopausal osteoporosis.
Radius Health is currently in arbitration proceedings with Ipsen in connection with the License Agreement. See “Legal Proceedings” for more information.
Eisai
In June 2006, the company exclusively licensed the worldwide rights to research, develop, manufacture and commercialize elacestrant and related products from Eisai (the "Eisai Agreement"). The company's license with Eisai did not originally include rights for Japan, however, on March 9, 2015, the company entered into an amendment to the Eisai Agreement in which Eisai granted it an exclusive right and license to research, develop, manufacture and commercialize elacestrant in Japan (the "Eisai Amendment"). Specifically, the company licensed the patent application that subsequently issued as U.S. Patent No. 7,612,114 (statutory term expires December 25, 2023 which the company expect will be extended to August 18, 2026 with 967 days of patent term adjustment due to delays by the USPTO), entitled "Selective Estrogen Receptor Modulator," the corresponding foreign patent applications and continuing patent applications. In consideration for the worldwide rights to elacestrant and in recognition of certain milestones having been met to date, Radius Health has paid to Eisai an aggregate amount of $1.9 million. Radius Health has also agreed to pay Eisai additional fees of up to $22.3 million, payable upon the achievement of certain clinical and regulatory milestones.
Under the Eisai Agreement as amended, should a product covered by the licensed technology be commercialized, the company will be obligated to pay to Eisai royalties in a variable mid-single digit range based on net sales of the product on a country-by-country basis. The royalty rate will be reduced, on a country-by-country basis, at such time as the last remaining valid claim in the licensed patents expires, lapses or is invalidated and the product is not covered by data protection clauses. In addition, the royalty rate will be reduced, on a country-by-country basis, if, in addition to the conditions specified in the previous sentence, lawful generic versions of such product account for more than a specified minimum percentage of the total sales of all products that contain the licensed compound during a calendar quarter. The latest licensed patent may expire, barring any patent term extension under any applicable laws, on August 18, 2026.
Radius Health was also granted the right to sublicense with prior written approval from Eisai. If the company sublicense the licensed technology to a third party, the company will be obligated to pay Eisai, in addition to the milestone fees referenced above, a fixed low double digit percentage of certain fees the company receive from such sublicensee in addition to royalties in the low single digit range based on net sales of the sublicensee.
The license agreement expires on a country-by-country basis on the later of (1) the date the last remaining valid claim in the licensed patents expires, lapses or is invalidated in that country, the product is not covered by data protection clauses, and the sales of lawful generic versions of the product account for more than a specified percentage of the total sales of all pharmaceutical products containing the licensed compound in that country, or (2) a period of 10 years after the first commercial sale of the licensed products in such country, unless it is sooner terminated.
The company can terminate the license agreement, with respect to the entire territory, with prior notice to Eisai if the company reasonably determine that the medical/scientific, technical, regulatory or commercial profile of the licensed product does not justify continued development or marketing.
Eisai can terminate the license agreement, on a country-by-country basis, at any time prior to the date on which Radius Health has submitted for either an NDA approval or EMA marketing approval with respect to a licensed product, upon prior written notice to it, if Eisai makes a good faith determination, in accordance with certain provisions specified in the agreement, that Radius Health has not used commercially reasonable efforts to develop the licensed product in the territory. Either party may also terminate the agreement upon an uncured material breach by the other party or upon the bankruptcy or insolvency of the other party. Eisai may terminate the license agreement, with prior written notice, in the event of certain changes of control involving it, if Eisai reasonably determines that the entity assuming control of it is not able to perform under the license agreement with the same degree of skill and diligence that the company would use. Eisai shall further have the right to terminate if the acquiring entity has any material and active litigations with Eisai or is a hostile takeover bidder against it.
Duke (RAD1901)
In December 2017, the Company entered into a License Agreement with Duke University (“Duke”) (collectively “Duke License Agreement”) pursuant to which Radius acquired the exclusive worldwide license to certain Duke patents associated with elacestrant (RAD1901) related to the use of elacestrant in the treatment of breast cancer as a monotherapy and in a combination therapy (collectively “Duke Patents”).
In consideration for these rights, the Company incurred non-refundable, non-creditable obligations to pay Duke, totaling $1.3 million, which were expensed as research and development during 2017. The Duke License Agreement provides for further payments upon the achievement of certain future regulatory and commercial milestones totaling up to $3.8 million. The agreement provides that the Company would pay Duke a fixed low single-digit royalty based on net sales, on a country-by-country basis, beginning in August 2029 and ending upon expiration of the last patent rights to expire. If the Company sublicenses the Duke Patents to a third party, the agreement provides that the Company will pay Duke a percentage of certain payments received by it from such sublicensee(s). The applicable percentage is in the high single-digit range on certain payments received in excess a pre-specified amount. The License Agreement may be terminated by Duke upon a material uncured breach of the License Agreement. The Company may terminate the License Agreement upon 60 days written notice.
Teijin Limited
In July 2017, the company entered into a license and development agreement with Teijin for abaloparatide-SC in Japan (the "Teijin Agreement"). Teijin is developing abaloparatide-SC in Japan under an agreement with Ipsen and has initiated a Phase 3 trial in Japanese patients with osteoporosis. Pursuant to the Teijin Agreement, the company granted Teijin ![]() an exclusive payment bearing license under certain of its intellectual property to develop and commercialize abaloparatide-SC in Japan, (ii) a non-exclusive payment bearing license under certain of its intellectual property to manufacture abaloparatide-SC for commercial supply in Japan, (iii) a right of reference to certain of its regulatory data related to abaloparatide-SC for purposes of developing, manufacturing and commercializing abaloparatide-SC in Japan, (iv) a manufacture transfer package, upon Teijin’s request, consisting of information and the Company’s know-how that is necessary for the manufacture of active pharmaceutical ingredient and abaloparatide-SC, (v) an obligation, at Teijin’s request, to manufacture (or arrange for a third party to manufacture) and supply (or arrange for a third party to supply) the active pharmaceutical ingredient for the clinical supply of abaloparatide-SC in sufficient quantities to enable Teijin to conduct its clinical trials in Japan, and (vi) an obligation, at Teijin’s request, to arrange for Teijin to directly enter into commercial supply agreements with the Company’s existing contract manufacturers on the same pricing terms and on substantially similar commercial terms to those set forth in the Company’s existing agreements with such contract manufacturers.
an exclusive payment bearing license under certain of its intellectual property to develop and commercialize abaloparatide-SC in Japan, (ii) a non-exclusive payment bearing license under certain of its intellectual property to manufacture abaloparatide-SC for commercial supply in Japan, (iii) a right of reference to certain of its regulatory data related to abaloparatide-SC for purposes of developing, manufacturing and commercializing abaloparatide-SC in Japan, (iv) a manufacture transfer package, upon Teijin’s request, consisting of information and the Company’s know-how that is necessary for the manufacture of active pharmaceutical ingredient and abaloparatide-SC, (v) an obligation, at Teijin’s request, to manufacture (or arrange for a third party to manufacture) and supply (or arrange for a third party to supply) the active pharmaceutical ingredient for the clinical supply of abaloparatide-SC in sufficient quantities to enable Teijin to conduct its clinical trials in Japan, and (vi) an obligation, at Teijin’s request, to arrange for Teijin to directly enter into commercial supply agreements with the Company’s existing contract manufacturers on the same pricing terms and on substantially similar commercial terms to those set forth in the Company’s existing agreements with such contract manufacturers.
In consideration for these rights, the company received an upfront payment of $10.0 million. The Teijin Agreement also provides for additional payments to it of up to an aggregate of $40.0 million upon the achievement of certain regulatory and sales milestones and requires Teijin to pay it a fixed low double-digit royalty based on net sales of abaloparatide-SC in Japan during the royalty term, as defined below. In addition, Radius Health has an option to negotiate a co-promotion agreement with Teijin for abaloparatide-SC in Japan.
The company maintain full global rights to its development program for abaloparatide-patch, which is not part of the Teijin Agreement. Pursuant to the Teijin Agreement, the parties may further collaborate on new indications for abaloparatide-SC.
Unless earlier terminated, the Teijin Agreement expires on the later of the ![]() date on which the use, sale or importation of abaloparatide-SC is no longer covered by a valid claim under its patent rights licensed to Teijin in Japan, (ii) expiration of marketing or data exclusivity for abaloparatide-SC in Japan, or (iii) 10th anniversary of the first commercial sale of abaloparatide-SC in Japan.
date on which the use, sale or importation of abaloparatide-SC is no longer covered by a valid claim under its patent rights licensed to Teijin in Japan, (ii) expiration of marketing or data exclusivity for abaloparatide-SC in Japan, or (iii) 10th anniversary of the first commercial sale of abaloparatide-SC in Japan.
Supply and Manufacturing Agreements
In June 2016, the company entered into a Supply Agreement with Ypsomed (the "Ypsomed Supply Agreement") pursuant to which Ypsomed agreed to supply commercial and clinical supplies of a disposable pen injection device customized for subcutaneous injection of abaloparatide (the "Device"). The company agreed to purchase a minimum number of Devices at prices per Device that decrease with an increase in quantity supplied. In addition, the company agreed to make milestone payments for Ypsomed’s capital developments in connection with the initiation of the commercial supply of the Device and to pay a one-time capacity fee. All costs and payments under the Ypsomed Supply Agreement are delineated in Swiss Francs.
The Ypsomed Supply Agreement has an initial term of three years from the earlier of the date of delivery of the first commercial batch of Devices after regulatory approval or June 1, 2017, after which, it automatically renews for two-year terms until terminated. The company or Ypsomed may terminate the agreement at any time by providing notice to the other party 18 months prior to the end of the then-current term. The agreement may also be terminated by either party upon material breach of the agreement, due to a party’s bankruptcy, insolvency, or dissolution, or due to a change of control of either party under certain circumstances. The company may terminate the agreement in the event that Ypsomed is unable to obtain regulatory or other approval for the manufacture and sale of Devices or if such approval is revoked. During the initial term of the agreement, the company estimate that the company will be obligated to make total minimum payments to Ypsomed of approximately CHF 3.9 million (approximately $4.0 million) in the aggregate, including the milestone payments and one-time capacity fee.
In June 2016, the company entered into a Commercial Supply Agreement (the "Vetter Supply Agreement") with Vetter Pharma International, GmbH ("Vetter") pursuant to which Vetter has agreed to formulate the finished abaloparatide-SC drug product containing the API of abaloparatide, to fill cartridges with the drug product, to assemble the pen delivery device, and to package and label the pen for commercial distribution. The company agreed to purchase the cartridges and pens in specified batch sizes at a price per unit. For labeling and packaging services, the company agreed to pay a per unit price dependent upon the number of pens loaded with cartridges that are labeled and packaged. These prices are subject to an annual price adjustment. The Vetter Supply Agreement has an initial term of five years, which began on January 1, 2016, after which, it automatically renews for two-year terms unless either party notifies the other party two years before the end of the then-current term that it does not intend to renew.
Vetter may terminate the Vetter Supply Agreement effective upon written notice to it if the company fail to maintain certain insurance required under the agreement, or breach provisions regarding ethical business practices, laws, and regulations. The company may terminate the agreement effective upon written notice to Vetter if: (1) Vetter fails to obtain or maintain any material governmental licenses or approvals, (2) Vetter has breached provisions regarding ethical business practices, laws, and regulations, or (3) the company fail to obtain certain regulatory approvals. Either party may terminate the agreement due to: (1) the other party’s bankruptcy or insolvency, (2) the other party’s uncured breach of the agreement, (3) a continuing force majeure event, or (4) a failure to reach mutual agreement on a change in the scope of work or services that Vetter reasonably believes it cannot perform because the change is in violation of applicable law.
In July 2016, the company entered into a Manufacturing Services Agreement (the "Manufacturing Agreement") with PPL, as successor-in-interest to Lonza, pursuant to which PPL has agreed to manufacture the commercial and clinical supplies of the API for abaloparatide. The company agreed to purchase the API in batches at a price per gram in euros, subject to an annual increase by PPL. Radius Health is also required to purchase a minimum number of batches annually beginning in 2018. The Manufacturing Agreement has an initial term of six years, after which, it automatically renews for three-year terms unless either party provides notice of non-renewal 24 months before the end of the then-current term.
PPL may terminate the agreement for any reason upon 30-months’ notice. The company may terminate the Manufacturing Agreement for any reason upon 24-months’ notice, if the company fail to obtain regulatory marketing approval for abaloparatide upon 12-months’ notice to PPL, or if abaloparatide is withdrawn from the market upon 12-months’ notice to PPL. Either party may terminate the agreement for the other's uncured breach of the agreement due to a party’s bankruptcy, insolvency, or dissolution, or due to certain force majeure events.




