Sesen Bio
Overview
Sesen Bio (SESN) (formerly Eleven Biotherapeutics) is a biologics oncology company focused primarily on designing, engineering and developing targeted protein therapeutics, or TPTs. The company's TPTs are single protein therapeutics composed of targeting moieties genetically fused via a peptide linker domain to cytotoxic protein payloads that are produced through its proprietary recombinant one-step manufacturing process. The company target tumor cell surface antigens that allow for rapid internalization into the targeted cancer cell and also have limited expression on normal cells. Sesen Bio has designed its TPTs to overcome the fundamental efficacy and safety challenges inherent in existing antibody drug conjugates, or ADCs, where a payload is chemically attached to a targeting antibody.
The company's most advanced product candidate is ViciniumTM, which is a locally-administered TPT. In a completed Phase 2 clinical trial, of the 45 evaluable subjects treated with Vicinium, 40% achieved a complete response or no evidence of disease at three months while 16% remained disease-free for at least 18 months. In the third quarter of 2015, Eleven Biotherapeutics ,through its subsidiary, Viventia Bio Inc., or Viventia, commenced in the United States and Canada a Phase 3 clinical trial of Vicinium for the treatment of subjects with high-grade non-muscle invasive bladder cancer, or NMIBC. The company completed enrollment in this clinical trial in March 2018 and anticipate reporting topline three-month data in mid-2018 and topline twelve-month data in the second quarter of 2019. In June 2017, the company entered into a Cooperative Research and Development Agreement, or CRADA, with the National Cancer Institute, or NCI, for the development of Vicinium in combination with AstraZeneca’s immune checkpoint inhibitor, durvalumab, for the treatment of NMIBC. Under the terms of the CRADA, the NCI will conduct a Phase 1 clinical trial in subjects with high-grade NMIBC to evaluate the safety, efficacy and biological correlates of Vicinium in combination with durvalumab. This Phase 1 trial is open and actively recruiting subjects.
The company's second most advanced product candidate is ProxiniumTM, a locally-administered TPT intended for the treatment of squamous cell carcinoma of the head and neck, or SCCHN. In its two Phase 1 clinical trials, 53% of evaluable subjects treated with Proxinium demonstrated antitumor activity with epithelial cell adhesion molecule, or EpCAM-expressing tumors as assessed by the investigators' clinical measurements, the investigators’ overall assessment including qualitative changes, and assessment of available radiologic data. In addition, three out of the four subjects with complete responses of injected tumors had regression or complete resolution of adjacent non injected lesions. In a Phase 2 clinical trial, the company observed tumor shrinkage in 10 of the 14 evaluable subjects (71.4%). The company intend to initiate a Phase 1/2a clinical trial that will explore the potential of Proxinium in combination with a checkpoint inhibitor for the treatment of SCCHN and are actively seeking partners for a combination program. In addition to its locally-administered TPTs, its pipeline also includes systemically-administered TPTs in development. The company's systemically-administered TPTs are built around its proprietary de-immunized variant of the plant-derived cytotoxin bouganin, or deBouganin. The company's lead systemically-administered product candidate, VB6-845d, is being developed for the treatment of multiple types of EpCAM-positive solid tumors. VB6-845d is administered by intravenous infusion. A Phase 1 clinical trial conducted with VB6-845, the prior version of VB6-845d, revealed no clinically relevant immune response to the deBouganin payload. The company plan on submitting an Investigational New Drug application, or IND, with VB6-845d, once funding or a partner is secured for this program.
Eleven Biotherapeutics has deferred further development of Proxinium and VB6-845d in order to focus its efforts and its resources on its ongoing development of Vicinium. Eleven Biotherapeutics is also exploring collaborations for Vicinium, Proxinium and VB6-845d.
The company maintain global development, marketing and commercialization rights for all of its TPT-based product candidates. The company intend to explore various commercialization strategies to market its approved products. If the company obtain regulatory approval for Vicinium in high-grade NMIBC, the company may build a North American specialty urology sales force to market the product or seek commercialization partners. If the company obtain regulatory approval for its other product candidates, including Proxinium, the company may seek partners with oncology expertise in order to maximize the commercial value of each asset or a portfolio of assets. The company also own or exclusively license worldwide intellectual property rights for all of its TPT-based product candidates, covering its key patents with protection ranging from 2018 to 2036. See ‘‘Business-Intellectual Property’’ for additional details.
On June 10, 2016, the company entered into the License Agreement with Roche, pursuant to which the company licensed its monoclonal antibody EBI-031 and all other IL-6 antagonist antibody technology owned by it. Under the License Agreement, Roche is required to continue developing EBI-031 at its cost. At the time of the License Agreement, EBI-031, which was derived using its previous AMP-Rx platform, was in pre-clinical development as an intravitreal injection for diabetic macular edema and uveitis. Eleven Biotherapeutics has received $30.0 million in payments from Roche pursuant to the License Agreement, including a $7.5 million upfront payment and a $22.5 million milestone payment as a result of the IND application for EBI-031 becoming effective. Eleven Biotherapeutics is also entitled to receive up to an additional $240.0 million upon the achievement of other specified regulatory, development and commercial milestones, as well as royalties based on net sales of potential future products containing EBI-031 or any other potential future products containing other IL-6 compounds.
The company also previously invested a significant portion of its efforts and financial resources in the development of its product candidate isunakinra (EBI-005) for the treatment of subjects with dry eye disease and allergic conjunctivitis. Based on negative results from its completed Phase 3 clinical trials in 2015 and 2016 for the treatment of dry eye disease and allergic conjunctivitis, the company do not plan to pursue further development of isunakinra.
TPT Platform
The company's current product candidates are based on its proprietary TPT platform and are focused on addressing areas of unmet medical need in cancer. The company's novel TPTs have been designed to overcome the efficacy and safety challenges of existing ADCs and are being developed for both local and systemic administration. The company's TPTs are single protein therapeutics composed of targeting moieties genetically fused via linker domains to cytotoxic protein payloads that are produced through its proprietary recombinant one-step manufacturing process. The company's TPT platform uses protein binding antibody fragments, which include fragment antigen binding domains, or Fabs, single chain variable domains, or scFvs, and non-covalent scFv dimers, or diabodies, derived from the domains of antibodies that confer antigen recognition. The company select antibody fragments for its product candidates depending upon the target therapeutic indication. The company target tumor cell surface antigens that allow for rapid internalization into the targeted cancer cell and that also have limited expression in normal cells. For local administrations, the company utilize an immunogenic cytotoxic protein payload designed to both target cancer cells and promote a heightened local immune response against the tumor. For systemic administrations, the company use a deBouganin payload that the company believe can be repeatedly administered via infusion without the generation of an efficacy-limiting immune response against the payload.
Locally-administered TPTs
The company utilize its TPTs with immunogenic cytotoxic protein payloads for tumors that can be targeted locally rather than systemically. Local administration allows for the TPT to reach the tumor without being cleared by the immune system, which enables it to maximize the concentration of TPTs directly to tumors. The company's locally-administered TPTs, including Vicinium, its lead product candidate, and Proxinium, contain a targeting moiety that is designed to bind to EpCAM, which is a protein over-expressed in many cancers. This targeting moiety is genetically fused to a truncated form of exotoxin A, or ETA, which is an immunogenic cytotoxic protein payload that is produced by the bacterial species, Pseudomonas. These product candidates are designed to bind to EpCAM on the surface of cancer cells. The TPT-EpCAM complex is subsequently internalized into the cell, and, once inside the cell, the TPT is cleaved by a cellular enzyme to release the cytotoxic protein payload, thus enabling cancer cell-killing.
The company also believe that its TPTs designed for local administration may not only directly kill cancer cells through a targeted delivery of a cytotoxic protein payload, but also potentiate an anti-cancer therapeutic immune response in cancer cells near the site of administration. This immune response is believed to be triggered by both the immunogenic cell death of the cancer cells due to its payload's mechanism of action and the subsequent release of tumor antigens and the immunologically active setting created by the nature of the cytotoxic protein payloads. The company believe that this immune response may also enhance the action of checkpoint inhibitors, which require a pre-existing immune response for maximum efficacy.
The company's most advanced locally-administered product candidates are Vicinium and Proxinium, for the treatment of high-grade NMIBC and recurrent, locally advanced or metastatic EpCAM-expressing SCCHN, respectively. These TPTs are not, however, suitable for systemic administration over multiple doses because the body’s immune system would recognize and eliminate foreign proteins, such as ETA, prior to their reaching targeted cancer cells.
Systemically-administered TPTs
The company also utilize its TPTs with a de-immunized payload where systemic administration is required. The company's systemically-administered TPTs currently in development are built around deBouganin. Since the body’s immune system naturally recognizes and attempts to eliminate foreign proteins, the company designed its systemically administered TPTs with a deBouganin payload to avoid inducing an immunogenic response. DeBouganin is constructed by mutating the immunogenic T-cell epitopes from bouganin so that they are not recognized as foreign by the immune system. However, the company also believe that deBouganin may enhance the action of checkpoint inhibitors as a result of the promotion of a local tumor immune response following the death of cancer cells. The company's systemically-administered product candidate is VB6-845d for the treatment of multiple types of EpCAM-positive solid tumors.
Differentiated Approach to Targeted Therapies
The company believe that its TPT construct will address many challenges experienced with existing ADCs. The basic construct for its TPTs and existing ADCs is similar as each is comprised of a targeting moiety that specifically binds to cancer cells and delivers a cytotoxic payload. However, existing ADCs have been associated with limitations that the company believe are addressed by its TPTs.
Limitations of existing ADC approaches to treating tumors
The company believe existing ADCs have the following fundamental efficacy and safety challenges:
Deliver insufficient drug to tumors. Existing ADCs utilize full-length antibodies, which, due to their large size, have a reduced ability to penetrate tumors, thereby reducing their efficacy.Inability to kill a broad array of cancer cells within a tumor. Subsets of cancer cells within tumors may have mechanisms to resist and not be responsive to the cytotoxic payloads, or small molecule chemotherapies, used in existing ADCs.Off-target toxicities due to unstable chemical linkage between targeting antibody and cytotoxic payload. Existing ADCs utilize chemical linkage strategies to join antibodies to small molecule cytotoxic payloads. While in the circulatory system, these chemical linkages can break and release free cytotoxic payloads in the circulation. These free small molecule cytotoxic payloads are not targeted and cannot discriminate between dividing cancer cells and non-cancerous cells, thus resulting in increased off-target toxicities.Limited combination therapy potential. The release of free cytotoxic payloads in the tumor region can result in toxicity to immune cells that attack tumors. This effect on anti-tumor immune cells may limit the potential utility of existing ADCs in combination therapies, including those employing immune checkpoint inhibitors.Complex and challenging manufacturing process. The multi-step manufacturing process of existing ADCs creates a non-homogeneous product that limits efficacy and drives greater costs than its manufacturing process.
Advantages of its TPT platform
The company believe its TPTs offer the following key advantages:
Deliver a greater amount of drug to tumors. The company's TPTs are designed using smaller targeting proteins that have an increased ability to exit the circulatory system and have binding properties designed to enable deeper penetration into targeted tumors, and the company believe this will increase efficacy.Ability to kill a broader array of cancer cells within a tumor. The company's novel cytotoxic payloads consist of proteins rather than small molecule cytotoxic payloads. The company believe the larger size of its cytotoxic protein payloads helps circumvent multi-drug resistance mechanisms that can make certain cancer cells resistant to small molecule cytotoxic payloads. By contrast to existing ADCs, which employ cytotoxic payloads that inhibit cellular replication and are effective at killing rapidly proliferating cancer cells, its cytotoxic protein payloads inhibit protein synthesis and are designed to kill not only rapidly proliferating, but also slowly growing cancer cells including tumor progenitor cells/cancer stem-like cells.Increase safety due to a more stable linkage between targeting protein and cytotoxic payload. The company's single protein molecules are designed to remain intact until they reach the inside of the cancer cell and to not release free cytotoxins into the circulatory system, thereby minimizing off-target toxicity.Promote a therapeutic immune response. The company believe that the potent TPT toxin-mediated killing of cancer cells in this immunologically active setting leads to the efficient presentation of cancer antigens to the immune system, thereby promoting an anti-tumor cellular immune response. The company's locally-administered TPTs utilize an immunogenic cytotoxic payload that the company believe promotes a heightened immune response in the local tumor environment.Potential combination with checkpoint inhibitors. The company believe that the potential effect of checkpoint inhibitors, which are antibodies that promote the action of anti-tumor T-cells by blocking inhibitory ligand/receptor interactions that include PD-1 and PD-L1, may be enhanced when used in combination with other agents. The company believe that, by mediating specific killing of tumor cells and promoting anti-tumor immune responses, its TPTs, while potentially effective on their own, may complement checkpoint inhibitors. In particular, the company believe that its use of its cytotoxin payload ETA, which promotes an immune response in the local tumor environment, may facilitate the presentation of tumor cell surface antigens following the death of cancer cells, thereby providing a tumor immune response to enhance the action of checkpoint inhibitor therapies.Utilize a simpler and more efficient manufacturing process. The company's proprietary recombinant one-step manufacturing process creates a homogeneous product that the company believe will improve efficacy and result in lower manufacturing costs.
Strategy
Eleven Biotherapeutics is committed to designing, engineering, developing and commercializing TPTs to identify and address oncology indications that suffer from a high unmet medical need. The key elements of its strategy are as follows:
Rapidly advance Vicinium through clinical development and obtain regulatory approval. Based upon its September 2014 end of Phase 2 meeting with the FDA, in the third quarter of 2015, Eleven Biotherapeutics ,through its subsidiary Viventia, commenced an open-label, non-randomized Phase 3 clinical trial of Vicinium in subjects with high-grade NMIBC in the United States and Canada. In November 2016, the FDA issued draft guidance regarding appropriate clinical trial design for new drugs and biologics for BCG-unresponsive NMIBC, including the use of single-arm studies. The FDA finalized this guidance in February 2018 and retained many of the recommendations from the 2016 draft guidance regarding clinical trial design, including the use of single-arm studies. The company believe that its Phase 3 clinical trial design is consistent with these aspects of the FDA’s guidance. The company completed enrollment of this Phase 3 clinical trial in March 2018 and anticipate reporting topline three-month data in mid-2018 and topline twelve-month data in the second quarter of 2019. If this Phase 3 clinical trial is successful, the company intend to pursue regulatory approval initially in the United States and Canada. Assuming that the company receive positive data in its Phase 3 clinical trial, the company intend to initiate discussions with the European Medicines Agency, or EMA, regarding a regulatory pathway for European Union, or E.U., approval.Explore opportunities in combination therapies. The company plan to continue discussions with potential partners that utilize technologies whose mechanism of action could be complementary to its TPT platform. These technologies include, but are not limited to, checkpoint inhibitors, immune modulators and other immuno-oncology agents. In June 2017, the company entered into a CRADA with the NCI for the development of Vicinium in combination with AstraZeneca’s immune checkpoint inhibitor, durvalumab, for the treatment of NMIBC. Under the terms of the CRADA, the NCI will conduct a Phase 1 clinical trial in subjects with high-grade NMIBC to evaluate the safety, efficacy and biological correlates of Vicinium in combination with durvalumab. This Phase 1 trial is open and actively recruiting subjects.Advance Proxinium through clinical development and obtain regulatory approval. The company intend to initiate a Phase 1/2a clinical trial that will explore the potential of Proxinium in combination with a checkpoint inhibitor. The company anticipate that the Phase 1/2a clinical trial will explore the potential for Proxinium, due to its potential immunogenic effect, to enhance checkpoint inhibitors in combination therapy for the treatment of EpCAM-expressing SCCHN.Advance its systemically-administered product candidate, VB6-845d. In April 2016, the company submitted an IND to the FDA in preparation of initiating a Phase 1/2 clinical trial of VB6-845d in subjects with EpCAM-positive cancers in the United States. The IND was withdrawn in July 2016 after the company received initial feedback from the FDA indicating that they had identified hold and non-hold deficiencies that needed to be addressed. In December 2016, the company submitted a request for a pre-IND meeting to seek input on the manufacturing, nonclinical and clinical plans for VB6-845d prior to resubmitting an IND. In February 2017, the FDA provided guidance on its manufacturing and nonclinical plans for VB6-845d. Based on this guidance, Eleven Biotherapeutics is performing additional studies to support an updated IND submission. The company believe that the deBouganin payload in VB6-845d may enhance the action of checkpoint inhibitors as a result of the promotion of a local tumor immune response following the death of cancer cells.Expand on the value of selected product candidates through strategic partnerships. The company may decide to selectively partner with pharmaceutical and biopharmaceutical companies when the company believe that a partner could bring additional resources and expertise to maximize the value of one or more of its product candidates.Leverage its TPT platform to develop additional product candidates. The company intend to develop additional product candidates based on its TPT platform. Depending on the strategic and financial merits, the company may enter into partnerships and collaborations to support these development efforts.Maximize the commercial value of its product candidates. The company maintain global development, marketing and commercialization rights for all of its TPT-based product candidates. If the company obtain regulatory approval for Vicinium in high-grade NMIBC, the company may build a North American specialty urology sales force to market the product in the United States and Canada or seek commercialization partners. Outside the United States and Canada, the company will seek commercialization partners with urology expertise. If the company obtain regulatory approval for its other product candidates, including Proxinium, the company may seek partners with oncology expertise in order to maximize the commercial value of each asset or a portfolio of assets.
Eleven Biotherapeutics has deferred further development of Proxinium and VB6-845d in order to focus its efforts and its resources on its ongoing development of Vicinium.
License Agreement with Roche
On June 10, 2016, the company entered into the License Agreement with Roche. Under the License Agreement, the company granted Roche an exclusive, worldwide license to develop and commercialize, at its cost, its monoclonal antibody EBI-031 and all other IL-6 antagonist antibody technology owned by it. Pursuant to the terms of the License Agreement, Roche is required to continue developing EBI-031 and any other product made from the other transferred IL-6 antagonist antibody technology at its cost.
Roche paid an upfront license fee of $7.5 million and a $22.5 million milestone payment as a result of the IND application for EBI-031 becoming effective. Roche has also agreed to pay up to an additional $240.0 million upon the achievement of specified regulatory, development and commercial milestones. In addition, Eleven Biotherapeutics is entitled to receive royalty payments in accordance with a tiered royalty rate scale, with rates ranging from 7.5% to 15% for net sales of potential future products containing EBI-031 and up to 50% of these rates for net sales of potential future products containing other IL-6 compounds, with each of the royalties subject to reduction under certain circumstances and to the buy-out options of Roche.
Product Pipeline
At this time Eleven Biotherapeutics is focused exclusively on the clinical development of Vicinium and have deferred further development of its other product candidates. The following table sets forth its current development stage programs:
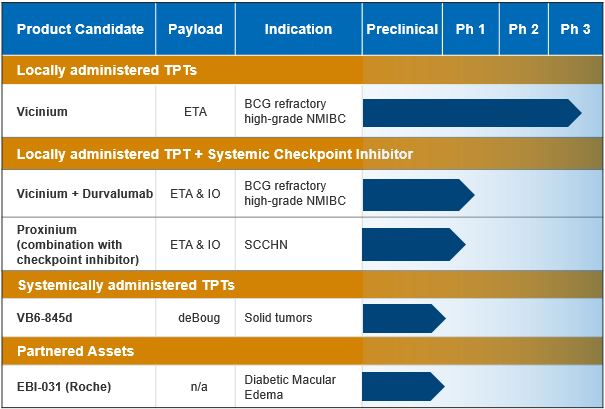
Vicinium
Overview
The company's most advanced locally-administered product candidate, Vicinium, is being developed for the treatment of high-grade NMIBC in subjects who have previously received two courses of BCG and whose disease is now BCG-unresponsive. Vicinium is administered by intravesical administration directly into the bladder. Vicinium utilizes an immunogenic cytotoxic protein payload that is a truncated form of ETA produced by the bacterial species, Pseudomonas. Vicinium also includes an anti-EpCAM ScFv targeting moiety that is required to deliver the ETA into EpCAM-expressing cancer cells. The toxicity to non-cancerous bladder cells is minimized due to their not having EpCAM over-expressed on their surface.
In a completed Phase 2 clinical trial, of the 45 evaluable subjects, 40% achieved a complete response, or CR, or no evidence of disease at three months while 16% remained disease-free for at least 18 months. Median time to disease recurrence was 274 days for subjects achieving a CR following a six-week induction phase, and this was extended to 408 days for subjects achieving a CR following a longer 12-week induction phase. No statistically significant difference was observed between the two dosing strategies (p=0.17). Vicinium was generally well-tolerated with no subjects discontinuing treatment in Phase 1 and Phase 2 clinical trials due to adverse events.
Based upon its September 2014 end of Phase 2 meeting with the FDA, Eleven Biotherapeutics ,through its subsidiary Viventia, commenced an open-label, non-randomized Phase 3 clinical trial of Vicinium in subjects with high-grade NMIBC who have received two courses of BCG, and whose disease is now BCG-unresponsive, and for whom the current standard of care is the surgical removal of their bladder, or a radical cystectomy, in the third quarter of 2015 in the United States and Canada. Based on safety and efficacy data observed with the longer 12-week induction in its Phase 2 clinical trial, the FDA agreed to its plan to employ more frequent dosing in its Phase 3 clinical trial, in which the primary end points are CR and duration of response, or DoR, in subjects with carcinoma in situ, or CIS, whose disease is BCG-unresponsive. In November 2016, the FDA issued draft guidance regarding appropriate clinical trial design for new drugs and biologics for BCG-unresponsive NMIBC, including the use of single-arm studies. The FDA finalized this guidance in February 2018 and retained many of the recommendations from the 2016 draft guidance regarding clinical trial design, including the use of single-arm studies. The company completed enrollment in this clinical trial in March 2018 and anticipate reporting topline three-month data in mid-2018 and topline twelve-month data in the second quarter of 2019. If this Phase 3 clinical trial is successful, the company intend to initially pursue regulatory approval in the United States and Canada.
As part of this trial, in July 2015, the company submitted a Clinical Trial Application, or CTA, to Health Canada to include Canadian sites. In September 2015, the company received a No Objection Letter from Health Canada, permitting it to proceed with its Phase 3 clinical trial in Canada. Assuming that the company receive positive data in its Phase 3 clinical trial, the company intend to initiate discussions with the EMA regarding a regulatory pathway for E.U. approval.
Overall, the company believe that its efficacy and safety data support the continued clinical development of Vicinium to fulfill a significant unmet medical need in subjects with high-grade NMIBC. Because Vicinium contains ETA, an immunogenic cytotoxic payload that elicits an anti-ETA immune response, the company believe the local administration of Vicinium may amplify the local host immune response within the tumor environment killing bladder cancer cells through an Immunogenic Cell Death, or ICD, mechanism. In addition, the company believe that this ICD response, which potentiates host immune responses against neoantigens present on the cancer cells, can lead to a heightened host immune response against their own tumor and potentially complement checkpoint inhibitor therapies.
The company own or exclusively license worldwide rights to its Vicinium intellectual property portfolio that provides unextended patent term until 2024, and, if its pending patent applications for Vicinium are granted patent protection until at least 2036. See ‘‘Business-Intellectual Property’’ for additional details.
Disease overview
Most cancers that form in the bladder are transitional cell carcinomas that derive from the transitional cell lining of the bladder. Transitional cell carcinoma of the bladder can be characterized as either high-grade or low-grade. Low-grade bladder cancer often recurs in the lining of the bladder after treatment, but rarely invades the muscular wall of the bladder or spreads to other parts of the body and is unlikely to be fatal. High-grade bladder cancer commonly recurs in the bladder, has a strong tendency to invade the muscular wall of the bladder, spreads to other parts of the body and is much more likely to result in death. Bladder cancer is also divided into muscle-invasive and NMIBC, based on invasion of the muscularis propria, which is the thick muscle deep in the bladder wall. Muscle invasive disease is more likely to spread to other parts of the body.
There are three forms of high-grade NMIBC, which are Ta, a papillary tumor in the innermost layer of the bladder lining, T1, a papillary tumor that has started to grow into the connective tissue beneath the bladder lining, and CIS, flat lesions of the transitional cell lining of the bladder. Papillary tumors are generally low-grade with low risk of progression, although about two to nine percent are high-grade, with a moderately high risk of progression to muscle-invasive bladder cancer. CIS tumors are always high-grade, with a worse prognosis than papillary tumors. CIS tumors appear irregular and abnormal under a microscope and the tumors are more aggressive, with a high probability of progression to muscle-invasive disease. Furthermore, the incidence of CIS in conjunction with Ta or T1 tumors results in a higher risk of recurrence and progression. About 75% to 85% of bladder cancers are non-muscle invasive. Of these, Ta tumors account for about 57% to 70%, CIS accounts for about 5% to 13% and T1 tumors account for about 20% to 30%.
Bladder cancer is the ninth most common cancer diagnosed worldwide and the second most common malignancy of the genitourinary system. There were an estimated 430,000 new cases of bladder cancer diagnosed in 2012 and 165,000 deaths worldwide. The global prevalence of bladder cancer is estimated at 2.7 million individuals. The American Cancer Society estimated that approximately 79,000 new cases of bladder cancer would be diagnosed in 2017 and there would be approximately 16,870 deaths due to bladder cancer in the United States during 2017. Based on a 2010 estimate prepared using Medicare data from the Surveillance, Epidemiology, and End Results program, among cancers in the United States, bladder cancer has the highest per-patient treatment costs, with an estimated overall cost of $3.9 billion annually. In the United States, bladder cancer has the highest overall cost among the elderly. Based on its assessment of the market, the treatment paradigm has remained the same since those figures were generated and the company believe the cost of care has increased.
NMIBC makes up 75% to 85% of all bladder cancers. The high recurrence rate and ongoing invasive monitoring requirement of bladder cancers are the key contributors to the economic and human toll of this disease. Bladder cancer occurs predominantly in older patients (about nine of the 10 people with bladder cancer are over the age of 55 years). The median age at diagnosis is approximately 73 years. Overall, the five year survival rate for bladder cancer in the United States is 77%. While the five year survival rates are 98% for stage zero and 88% for stage one NMIBC, once the cancer becomes invasive, the rates drop dramatically with five year survival rates of 63%, 46% and 15% for stage two, three and four muscle invasive bladder cancers, respectively. Eleven Biotherapeutics is targeting subjects with BCG-unresponsive high-grade NMIBC. The company's initial target market includes the approximately 25,000 patients diagnosed annually, as well as the patients who have previously failed BCG and have refused cystectomy. The company would expect that, if Vicinium is approved by the FDA, patients would receive treatment until the earlier of 2 years and disease recurrence.
Current approaches to treatment
Within high-grade NMIBC, the initial treatment of Ta or T1 is transurethral resection of the bladder tumor, or TURBT, followed by BCG treatment. For CIS, whether or not TURBT is an option, BCG is the standard of care. BCG is a live attenuated strain of Mycobacterium bovis, with a diminished virulence in humans. Since BCG works by utilizing an immune/inflammatory mechanism, BCG is generally initiated only two to four weeks after TURBT, allowing the urothelium to heal and lowering the risk of systemic infection. When high-grade bladder tumors have been completely resected, BCG is used as adjuvant therapy to prevent recurrence. In patients with residual disease after resection, BCG helps to eradicate residual disease and delay progression. The BCG regimen consists of an induction phase followed by a maintenance phase. The induction phase involves six consecutive once-weekly instillations of the drug into the bladder. The maintenance phase involves three consecutive once-weekly instillations repeated every three to six months for at least one year. The response rate to a single induction phase of BCG is 60% to 70% with an additional 30% to 50% of the non-responders becoming responders following a second induction phase. However, BCG’s failure rate for all responders is estimated to be as high as 50% within the first 12 months of treatment and 90% within five years.
For patients who received BCG, and whose disease is now BCG-unresponsive, radical cystectomy is recommended due to the risk of progression to muscle invasive disease, which greatly reduces a patient’s prognosis. Radical cystectomy is a complex surgery associated with a significant morbidity rate of 28% to 45% and a mortality rate of 8% within six months of surgery. The surgery also entails a number of short-term risks including bleeding and/or clots, infections, bowel obstruction, bowel perforation, peritonitis and injury to the urethra. More than 25% of radical cystectomy patients require readmission for surgery-related complications within 90 days following surgery, and 34% require emergency room visits. The impact of radical cystectomy is life-altering, with major lifestyle changes, including incontinence and sexual dysfunction, and daily issues related to management of the external bag for urine collection.
In 2009, Valstar was re-launched in the United States for the treatment of BCG-refractory CIS bladder cancer in patients for whom radical cystectomy is not an option. Valstar is administered intravesically directly into the bladder once a week for six weeks. Due to drug resistance and toxicities, Valstar has had limited utility. Other than Valstar, there are no other approved therapies for CIS bladder cancer. However, there are various other product candidates in development for the treatment of NMIBC, including product candidates developed by AADi, LLC (ABI-009), Altor BioScience Corp. (ALT-801), Cold Genesys, Inc. (CG0070) and FKD Therapies Oy (Instiladrin). Clinical trials and pre-clinical studies
Pre-clinical studies. The company's in vitro studies of Vicinium in bladder cancer cell lines demonstrated activity following an exposure time equivalent to clinical dosing. The anti-tumor activity of Vicinium was also evaluated against human tumor xenografts (SCCHN, colorectal and small cell lung carcinoma cell lines) using an athymic mouse model. Mice bearing EpCAM-positive human tumor xenografts implanted subcutaneously were administered 0.25-0.5 mg/kg of Vicinium by intravenous injection, and tumor size monitored over the course of the pre-clinical study (33 to 51 days post-initiation of treatment) and compared to that of an untreated tumor-bearing group. Vicinium demonstrated significant tumor growth suppression. Vicinium is designed to be a local therapy and is administered by intravesical instillation directly to the bladder. Vicinium repeatedly administered subcutaneously in both rats and cynomolgus monkeys did not result in any product candidate-related systemic toxicity. Toxicities associated with subcutaneous administration were limited to localized irritation with skin lesions resolving by the end of the pre-clinical study. Vicinium was found to be immunogenic in all species tested with anti-drug antibodies observed after seven days of dosing.
Phase 1 clinical trial. The company initiated an open-label, dose-escalating Phase 1 clinical trial of Vicinium in September 2004 at 22 sites in Canada. The company enrolled 64 subjects with high-grade Ta or T1 tumors with or without CIS (17 of which had CIS) and who had previously received at least one treatment of BCG. The Phase 1 clinical trial was designed to assess safety and determine the maximum tolerated dose, and the recommended Phase 2 dose. The secondary objective was to explore the anti-tumor activity of Vicinium.
Eight dose levels were initially evaluated, ranging from 0.1 to 10.56 mg dose given once weekly for six consecutive weeks. Each dose was administered by instillation and held for two hours prior to voiding. Safety data from each dose cohort was evaluated after three weeks of treatment before proceeding to the next dose cohort. A maximum tolerated dose was not reached; therefore, additional escalations through 13.73 mg, 17.85 mg, 23.20 mg and 30.16 mg were undertaken. No dose-limiting toxicities, or DLTs, were reported and no maximum tolerated dose was reached in these additional dose-escalations. Vicinium was generally well-tolerated at each of these escalated doses.
A CR was defined in this Phase 1 clinical trial as non-positive urinary cytology and either normal cystoscopy or abnormal cystoscopy with negative biopsy. Of the 64 subjects enrolled, only 61 were considered to be evaluable for efficacy as two subjects were excluded from the analysis due to an absence of BCG treatment prior to this Phase 1 clinical trial, and there was one unrelated death for whom no final tumor assessment was obtained. Evidence of clinical efficacy, as defined by a CR, was achieved by 24 of the 61 randomized subjects (39%). Only three of the 17 subjects (18%) treated in the 0.1-<1mg/dose range were CRs. In contrast, seven of the 14 subjects (50%) treated in 1.0-<10mg/dose range and 14 of the 30 subjects (46.7%) treated in the ≥10mg/dose range experienced CRs at the three month assessment. Of the subjects with CIS, five of the 17 subjects (29%) achieved a CR, while non-recurrence was observed in seven of the 16 subjects with T1 (43.8%) and 12 of the 28 subjects with Ta (42.8%). This Phase 1 clinical trial was completed in April 2006.
Phase 2 clinical trial. Based on its Phase 1 clinical trial conducted in Canada, the company submitted the IND for Vicinium to the FDA in August 2005, and the company initiated an open-label Phase 2 clinical trial of Vicinium in March 2007 at 20 sites, in Canada and the United States. The company enrolled 46 subjects with CIS (with or without Ta or T1) who had previously received at least one treatment of BCG. Of the 46 subjects enrolled, 27 subjects (58.7%) had received at least two treatments of BCG. The Phase 2 clinical trial was designed to determine the tolerability and explore the potential for clinical benefit from Vicinium. Clinical benefit was defined in this Phase 2 clinical trial as a CR or no evidence of disease at the three month evaluation. A CR was defined in this Phase 2 clinical trial as no histological evidence of disease and negative urine cytology. Any cases with no histological evidence of disease on initial biopsy but atypical or suspicious urine cytology were also considered CRs only if they remained negative after being evaluated with repeat biopsy, directed and random. Vicinium showed evidence of clinical efficacy. A subject was considered to have a durable CR if that subject obtained a CR and remained disease-free for a period of at least 12 months from initiation of treatment.
The dosing regimen for its Phase 2 clinical trial included an induction phase followed by a maintenance phase, consisting of three weekly treatments and then nine weeks of no treatment repeated every three months for at least one year and ending with nine weeks of no treatment. There were two treatment groups in this Phase 2 clinical trial. Treatment Arm A consisted of 23 subjects, of which 22 were ultimately evaluable as one subject violated eligibility requirements early in this Phase 2 clinical trial. Twenty-two subjects in the induction phase received six consecutive once-weekly instillations of 30 mg of Vicinium. At the three-month assessment, subjects with residual disease but no disease progression-where disease progression is defined as being muscle invasive-were eligible for either a second induction phase or a maintenance phase, which consisted of three consecutive once-weekly instillations repeated every three months for at least one year. Of the 13 subjects unable to achieve a CR at the three-month assessment, nine subjects elected additional treatment. From these nine, two became CRs after receiving maintenance dosing. Treatment Arm B was added to evaluate a longer induction cycle. In Treatment Arm B, 23 subjects in the induction phase received 12 consecutive once-weekly instillations of 30 mg Vicinium. At the three-month assessment, the CR rate for both treatment arms was 40%. At the 12-month assessment, the CR rate in Treatment Arm A was 13%, but 17% in Treatment Arm B. Of those subjects who did not achieve a CR at the three-month assessment, 73% had either a reduction in tumor size or did not experience further tumor growth. The following charts demonstrate the responses in this Phase 2 clinical trial in Treatment Arm A and Treatment Arm B. The data below shows the percentage change in surface area of cancer within the bladder, based on bladder mapping data utilizing cystoscopy in 40 subjects.


This Phase 2 clinical trial was completed in September 2009.
Near the completion of this Phase 2 clinical trial in 2009, Valstar was re-launched in the United States for the treatment of BCG-refractory CIS bladder cancer in subjects for whom immediate cystectomy would be associated with unacceptable morbidity or mortality. However, because physicians were not widely prescribing Valstar to their patients and it was not an approved therapy in Europe, this disrupted its originally designed clinical path of a head-to-head pivotal Phase 3 clinical trial of Vicinium against Valstar. Due to the uncertainty of the standard of care in this space, its efforts were put on hold until a clear clinical path was established. In May 2013, the FDA co-sponsored a public workshop where it evaluated potential trial designs for the development of therapies for NMIBC and specifically provided regulatory guidance supporting the idea that a single-arm clinical trial could provide sufficient evidence of benefit if the results were robust. The panel suggested it is acceptable to include high-grade papillary subjects without CIS in a clinical trial with CIS subjects because the clinical management and outcome if left untreated is considered to be the same. Thereafter, the company began discussions with the FDA and refocused its resources to commence an open-label, non-randomized Phase 3 clinical trial of Vicinium in subjects with high-grade NMIBC.
Safety data. The company believe that its safety data from 110 subjects in its Phase 1 and Phase 2 clinical trials support further development of Vicinium. There were no Grade 4 or Grade 5 serious adverse events that were considered by the clinical investigators to be related to Vicinium. There was one Grade 5 serious adverse event, or death, which was determined by the clinical investigator to be unrelated to Vicinium. The most common treatment-related adverse events were an abnormally frequent passage of small amounts of urine, blood in the urine and painful urination, the majority of which were considered to be mild or moderate in severity. No subjects discontinued treatment due to a Vicinium-related adverse event during the Phase 1 and Phase 2 clinical trials.
Vicinium Phase 3 clinical trial development plan
Based upon its September 2014 end of Phase 2 meeting with the FDA, Eleven Biotherapeutics ,through its subsidiary Viventia, commenced an open-label, non-randomized Phase 3 clinical trial of Vicinium in subjects with high-grade NMIBC who have received two courses of BCG, and whose disease is now BCG-unresponsive, and for whom the current standard of care is the surgical removal of their bladder, or a radical cystectomy, in the third quarter of 2015 in the United States and Canada. Based on safety and efficacy data observed with the longer 12-week induction in its Phase 2 clinical trial, the FDA agreed to its plan to employ more frequent dosing in its Phase 3 clinical trial, in which the primary end points are CR and DoR in subjects with CIS whose disease is BCG-unresponsive. In November 2016, the FDA provided draft guidance regarding appropriate clinical trial design for new therapies for NMIBC, including the use of single-arm studies, and the company believe that its Phase 3 clinical trial design is consistent with the FDA’s draft guidance. The company completed enrollment in this clinical trial in March 2018 and anticipate reporting three-month topline data in mid-2018 and topline twelve-month data in the second quarter of 2019. If this Phase 3 clinical trial is successful, the company intend to initially pursue regulatory approval in the United States and Canada.
As part of this trial, in July 2015, the company submitted a Clinical Trial Application, or CTA, to Health Canada to include Canadian sites. In September 2015, the company received a No Objection Letter from Health Canada, permitting it to proceed with its Phase 3 clinical trial in Canada. Assuming that the company receive positive data in its Phase 3 clinical trial, the company intend to initiate discussions with the EMA regarding a regulatory pathway for E.U. approval.
Proxinium
Overview
The company's second most advanced locally-administered product candidate, Proxinium, is being developed as a treatment for subjects with recurrent or metastatic EpCAM-expressing SCCHN who have received at least one prior platinum-based chemotherapy regimen with recurrent, locally advanced or metastatic EpCAM-expressing SCCHN. Proxinium utilizes an immunogenic cytotoxic protein payload that is a truncated form of ETA produced by the bacterial, Pseudomonas, and is designed to target EpCAM-positive cancer cells, while minimizing toxicity to non-cancerous cells. Proxinium is administered via injection directly into the targeted tumor, or intratumoral injection. Proxinium has received Orphan Drug Designation from the FDA and the EMA and Fast Track designation from the FDA.
In its two Phase 1 clinical trials, subjects treated with Proxinium had demonstrated antiitumor activity in 53% of evaluable subjects with EpCAM-expressing tumors as assessed by the investigators’ clinical measurements, the investigators' overall assessment including qualitative changes, and assessment of available radiologic data. In addition, three out of the four subjects with complete responses of injected tumors had regression or complete resolution of adjacent non injected lesions. In a Phase 2 clinical trial, the company observed tumor shrinkage in 10 of the 14 evaluable subjects (71.4%). Combined results from two Phase 1 clinical trials encompassing 44 subjects have shown complete resolution or reduction in size of injected tumors in 16 of the 30 evaluable EpCAM-positive subjects (53.3%). An additional 27% of evaluable subjects had stable disease and, therefore, the results indicate an overall tumor control rate of approximately 80%. Proxinium was generally well-tolerated during the clinical trials. Dose-limiting toxicity in the Phase 1 clinical trials was transaminase elevation in liver enzymes.
In its clinical trials involving Proxinium, Eleven Biotherapeutics has also observed some stabilization, partial reduction and complete resolution of non-injected tumors. The company believe that TPT mediated killing of cancer cells occurs via a mechanism known as ICD, which is known to enhance the presentation of neoantigens to the immune system. The company believe that this, combined with the immunogenic nature of its cytotoxic protein payload creates a heightened immune response, wherein naive cytotoxic T cells are stimulated by antigen presenting cells, such as dendritic cells, presenting tumor cell surface antigens following the death of cancer cells. The company believe that this anti-tumor response may complement checkpoint inhibitor therapies.
The company intend to initiate a Phase 1/2a clinical trial that will explore the potential of Proxinium in combination with a checkpoint inhibitor for the treatment of SCCHN and are actively seeking partners for a combination program. The company anticipate that the Phase 1/2a clinical trial will explore the potential for Proxinium, due to its potential immunogenic effect, to enhance checkpoint inhibitors in combination therapy for the treatment of SCCHN. The company will be measuring both the objective response rates and immune response biomarkers in a Phase 1/2a clinical trial. Should a trial yield encouraging results and Eleven Biotherapeutics is able to secure additional funding, the company will move into later stage trials.
During a Type C meeting with FDA in 2007, the FDA noted that approval of a companion diagnostic for EpCAM expression would need to coincide with Proxinium approval. During the clinical evaluation of Proxinium, the company developed an immunohistochemical test to determine whether clinical trial subjects are EpCAM-positive. Internal examination from head and neck cancer subjects showed that its EpCAM antibody bound to 84% of all subject samples. The company intend to seek the FDA’s input as to whether this immunohistochemical test satisfies the FDA’s request for a companion diagnostic for EpCAM expression and whether the company will need to submit this test for pre-market approval as a companion diagnostic in conjunction with Proxinium.
Overall, the company believe that its efficacy and safety data support the continued clinical development of Proxinium to fulfill a significant unmet medical need in subjects with recurrent, locally advanced or metastatic EpCAM-expressing SCCHN.
The company believe the local administration of Proxinium mediates ICD of cancer cells leading to the release of tumor-specific neoantigens and attracting/activating cells of the host immune system. Further, Proxinium, like Vicinium, contains ETA, an immunogenic cytotoxic payload. The local activation of an anti-ETA response may further heighten the local immune response. The company also believe that the effect of checkpoint inhibitors may be enhanced if they are used in combination with Proxinium due to its potential immunogenic effect.
Eleven Biotherapeutics has deferred further development of Proxinium in order to focus its efforts and its resources on its ongoing development of Vicinium. Eleven Biotherapeutics is also exploring collaborations for Proxinium.
The company own or exclusively license worldwide rights to its Proxinium intellectual property portfolio that provide unextended patent term until 2024 and, if its pending composition of matter patent applications for Proxinium are granted, until at least 2036. See ‘‘Business-Intellectual Property’’ for additional details.
Disease overview
Head and neck cancers, which include cancers of the oral cavity, pharynx and larynx, are collectively the sixth most common cancers in the world. Head and neck cancer develops from the mucosal linings of the upper aerodigestive tract, comprising of the nasal cavity and paranasal sinuses, the nasopharynx, the hypopharynx, larynx, and trachea, and the oral cavity and oropharynx. Squamous cell carcinoma is the most frequent malignant tumor of the head and neck region. Invasive head and neck cancers arise in most cases from preneoplastic lesions grouped under the term dysplasia. Dysplastic lesions present with an increased likelihood of progressing to squamous cell carcinoma. The altered epithelium displays architectural and cytological changes that range from mild to severe.
There are approximately 650,000 new cases annually and nearly 350,000 deaths due to head and neck cancer. In head and neck cancer, approximately 40% to 60% of deaths result from local or regional disease. The American Cancer Society estimates that there will be approximately 64,690 new cases of head and neck cancers in the United States in 2018, of which 51,540 cases would be attributed to cancers of the oral cavity and pharynx and 13,150 cases would be attributed to cancer of the larynx, and 13,740 deaths. Most of head and neck cancers are biologically similar with more than 90% being squamous cell carcinomas that commonly originate in the epithelium. They are strongly associated, more so than other cancers, with certain environmental and lifestyle risk factors, and worldwide incidence exceeds 650,000 cases annually.
New treatment modalities have been undermined by the approximately 10% to 25% of cured patients who subsequently develop second primary tumors due to field cancerization. These second primary tumors are one of the leading factors in the 40% to 50% five year survival rate. Based on its immunohistochemical test used during its clinical trials of Proxinium, the company believe that approximately 84% of late-stage SCCHN are EpCAM-positive. Eleven Biotherapeutics is initially developing Proxinium to potentially address late-stage SCCHN. The company would expect that, if Proxinium is approved by the FDA, patients would receive treatment until disease progression.
Current approaches to treatment
Existing treatment options for SCCHN include immunotherapy (checkpoint inhibitors), surgery, drug therapies, radiation therapy or a combination of therapies. There is no standard treatment option for patients who progress after receiving these treatments. Approximately 60% of patients with head and neck cancer have locally recurrent disease and distant metastases occur in 20% to 30% of patients.
Currently, Erbitux, an anti-epidermal growth factor receptor antibody, is the only FDA approved tumor-targeted therapy for the treatment of late-stage SCCHN. Erbitux has been approved as a first-line therapy for late-stage SCCHN in combination with platinum-based therapy plus fluorouracil. Erbitux has also attained approval as a monotherapy or in combination with radiation therapy for second-line treatment of late-stage SCCHN in patients that have failed platinum-based therapy.
The five year survival rate for patients with locally recurrent disease is reported to be 40% to 50% and loco-regional recurrence is the predominant cause of failure and up to 70% of such patients have advanced disease. In addition, more than 50% of all patients who die from head and neck cancers have loco-regional disease as the only site of failure. If head and neck cancers are not controlled locally, they can infringe on the esophagus and airway, compromising nutrition and respiratory functions and often resulting in significant anatomic disfigurement. As such, the management of locally recurrent disease requires a multidisciplinary approach involving an array of specialists with an expertise in head and neck cancers.
Most recently checkpoint inhibitors have entered into use in the treatment of SCCHN. Two checkpoint inhibitors that target PD-1, pembrolizumab and nivolumab, have now received approval for the treatment of SCCHN. Nivolumab was granted FDA approval for the treatment of patients with SCCHN who have progressed on or after platinum-based chemotherapy. Nivolumab-treated subjects had a 30% reduction in the risk of death; a median OS of 7.5 months for nivolumab and 5.1 months for investigator's choice, or IC. There were no statistically significant differences between the two arms for progression-free survival or objective response rate, or ORR (13.3% versus 5.8% for nivolumab and IC, respectively). Pembrolizumab was granted accelerated approval by the FDA for the treatment of patients with SCCHN who have progressed on or after platinum-based chemotherapy. The major efficacy outcome measures were ORR according to Response Evaluation Criteria in Solid Tumors, or RECIST version 1.1, as assessed by blinded independent central review, and duration of response. The ORR was 16% (95% CI: 11, 22) with a complete response rate of 5%.
Clinical trials and pre-clinical studies
Pre-clinical studies. Pre-clinical data from in vitro and in vivo studies of Proxinium have demonstrated the potential to be safe in humans and to have clinical activity. In vitro pharmacology studies have demonstrated that Proxinium exhibits potent cytotoxicity against numerous EpCAM-positive cell lines, including SCCHN, bladder tumor and prostate tumor cell lines. Proxinium has also demonstrated anti-tumor activity in several human tumor xenograft animal models expressing EpCAM, including those derived from human squamous cell carcinomas and in a lung cancer subject derived xenograft, or PDX, model. In the PDX model, mice engrafted with human bone marrow cells were each implanted with two subcutaneous EpCAM-positive human PDX tumors. Tumor-bearing animals were treated with Proxinium alone in one tumor, an anti-PD-1 checkpoint inhibitor alone given systemically, or a combination of the two. Intratumoral injection of Proxinium into the tumor xenograft located on one side of the animal was able to block the growth of the tumor while also having a quantifiable anti-tumor effect on the uninjected tumor on the opposite flank. In contrast, the checkpoint inhibitor alone had little effect on tumor growth but appeared to amplify Proxinium’s activity on the uninjected tumor.


Proxinium treatment alone was also observed to increase the numbers of CD8+ T cells, an important cytotoxic immune cell population, in the blood. The company believe that this indicates that the checkpoint inhibitor alone was ineffective in initiating the anti-tumor effects of immune cells without the prior activation of cancer specific immune cells by a cytotoxic regimen, such as Proxinium.
Further, synergistic and additive effects were observed in these in vitro pharmacology studies with Proxinium in combination with various anti-cancer agents, as well as with radiation therapy. Toxicological studies in Sprague-Dawley rats showed no clear evidence of systemic toxicity whether given via intradermal or subcutaneous injection. The only dose-related reactions were at the injection site with most lesions resolving by the end of the observation period. Plasma concentrations of animals in a seven-day toxicology study conducted in Sprague-Dawley rats were 50 ng/mL at four hours after squamous cell injection and approximately 1,000 ng/mL at 10 minutes following intravenous injection on day one. These blood levels were approximately 5- and 100-fold higher, respectively, than the mean Cmax measured in subjects administered 700 mg weekly for four weeks (10,936 pg/mL) through the intratumoral route. In in vivo pharmacology studies in human tumor xenograft mouse models, the company observed evidence of tumor growth suppression.
In summary, in vitro and in vivo pre-clinical data have shown that the anti-cancer agent Proxinium preferentially binds to tumor cells and has an acceptable safety profile. The local and systemic toxicological profile for Proxinium in Sprague-Dawley rats has been defined with toxicological effects at doses 1,000-fold greater than the IC50 for activity on tumor cells.
First Phase 1 clinical trial. The company initiated an open-label, dose-escalating Phase 1 clinical trial of Proxinium in December 2003 at three sites in Russia. The company enrolled 24 subjects with late-stage SCCHN who had previously undergone prior radiation therapy, with a majority having completed at least one chemotherapy cycle. In addition, based on its immunohistochemical test used during this Phase 1 clinical trial of Proxinium, 17 of the 24 subjects (70.8%) enrolled in this Phase 1 clinical trial had tumors that tested positive for EpCAM. The Phase 1 clinical trial was designed to determine the maximum tolerated dose and the recommended Phase 2 dose. Secondary objectives included evaluation of safety, tolerability, PK profile and efficacy endpoints.
In addition, information on PK properties and immunogenicity, as well as a preliminary assessment of tumor response, was obtained.
Subjects received two cycles of Proxinium administered once per day for five consecutive days, with doses ranging from 20 µg to 280 µg, followed by 23 days off. Two DLTs occurred at the 280 µg dose level, establishing 200 µg as the maximum tolerated dose. The DLTs observed in the two subjects were elevated liver enzyme tests, which were not associated with any signs of liver damage or toxicity, were asymptomatic and were transient as they resolved to baseline values.
Objective anti-tumor responses were measured by caliper, CT scans and digital photography from baseline to final assessment. Anti-tumor responses and stable disease were observed in six of the 14 (42.9%) and four of the 14 (28.6%) evaluable subjects with EpCAM-positive tumors, respectively, for an overall response rate of 71.4% (10 of the 14 evaluable subjects). All six of the evaluable subjects with EpCAM-negative tumors had progression of their target tumors. In addition, the 10 subjects with EpCAM-positive tumors and who had responses or stable disease had a survival time of 278 days compared with a survival time of 124 days for the six subjects with EpCAM-negative tumors. The 14 evaluable EpCAM-positive subjects had a survival time of 207 days. This Phase 1 clinical trial was completed in October 2004.
Second Phase 1 clinical trial. The company initiated a second open-label, dose-escalating Phase 1 clinical trial of Proxinium in June 2004 at four sites in Brazil. The company enrolled 20 subjects with late-stage SCCHN. All of the subjects enrolled in this Phase 1 clinical trial had undergone prior radiation therapy, with a majority having completed at least one chemotherapy cycle. Eighteen of the 20 subjects (90%) tested positive for EpCAM, of which two subjects were non-clinically evaluable. Preliminary efficacy data indicated 14 of the 16 evaluable EpCAM-positive subjects (87.5%) had either a ‘‘complete resolution,’’ ‘‘response,’’ or ‘‘stable’’ disease of injected tumors following Proxinium treatment, with 25% of subjects achieving a ‘‘complete response’’ of the injected tumor. The second Phase 1 clinical trial was designed to determine the maximum tolerated dose and the recommended Phase 2 dose. Secondary objectives included evaluation of safety, tolerability, PK profile and efficacy endpoints.
Subjects received Proxinium once weekly for four weeks with initial doses ranging from 100 µg to 930 µg, followed up by four weeks with once weekly doses ranging from 100 µg to 930 µg. The maximum tolerated dose was established at 700 µg, based on the occurrence of DLTs in two of the five subjects at the 930 µg dose level cohort. The DLTs observed in the two subjects were elevated liver enzyme tests. In both cases, the elevated liver enzyme tests were not associated with any signs of liver damage or toxicity, were asymptomatic, and resolved to baseline values.
Therapeutic exploratory endpoints were analyzed to evaluate the tumor response and anti-tumor activity of Proxinium. RECIST criteria were not used in this Phase 1 clinical trial and instead the clinical investigator used the following definitions for tumor responses: ‘‘complete response’’ was the complete clinical resolution of a tumor (injected or non injected), ‘‘response’’ was defined as clinically and radiologically documented reduction in the size of the target tumor indicative of anti-tumor activity from baseline to final, ‘‘stable’’ was defined as no change in the disease state captured through clinical or radiological assessments from baseline to final and ‘‘progression’’ was defined as an increase in the size of the target tumor from baseline to the final assessment.
A non injected tumor response was observed in four of the 20 subjects (20%) in the second Phase 1 clinical trial where subjects were administered Proxinium weekly and one of the 15 subjects (6.7%) in the Phase 2 clinical trial, which is discussed below, where subjects were administered Proxinium weekly. In such cases, tumor responses were seen in loco-regional tumors that themselves had not been injected with Proxinium, but were adjacent to, and in one case bi-lateral to the injected tumor. An example of a non-target tumor response is shown in FIGURE 3 and FIGURE 4 above. The company believe that this non-target tumor response may be a consequence of surrounding cancer cells dying in response to Proxinium through diffusion (or the local spread of Proxinium) and cross priming of the immune system or the selective release of cancer antigens into the local immune tumor environment. This Phase 1 clinical trial was completed in March 2005.
Phase 2 clinical trial. The company submitted the IND for Proxinium to the FDA in August 2005. The company initiated an open-label Phase 2 clinical trial of Proxinium in March 2006 at nine sites in Canada and twenty-one sites in the United States. The company enrolled 15 subjects with refractory locally recurrent disease, which means that the subject must have progressed on or after receiving, or was unable to tolerate, at least one anti-cancer treatment regimen or had to have previously documented refusal of treatment for locally recurrent disease. The Phase 2 clinical trial was designed to determine the safety, tolerability and recommended dose of intratumorally injected Proxinium. Secondary objectives were to evaluate principal target tumor and target tumor response rates, determine the time to progression of the principal target tumor, overall survival time and progression-free survival, and also to confirm the PK profile and assess immunogenicity of intratumorally injected Proxinium.
The dosing regimen for its Phase 2 clinical trial included intratumoral injections once per week at 500µg or 700µg. Although the small sample size does not allow for statistical evaluation of treatment effects, it was expected that the Phase 2 clinical trial would provide some additional evidence of the therapeutic effect of Proxinium for control of local or regional late-stage SCCHN, as well as a survival benefit for those subjects. To be eligible for measurement of a radiographically confirmed response, a subject had to have a radiographic assessment at baseline/day one and at least two additional radiographic assessments, not less than four weeks apart, after day one. According to this definition, eight subjects were eligible for a principal tumor radiological response evaluation. In order to assess best tumor response, at least two CT scans, one at baseline and one post-baseline, were required. Best tumor response was defined in this Phase 2 clinical trial as the greatest degree of decrease in tumor size observed throughout the clinical trial. Bidimensional tumor measurements were used to determine tumor size. Bidimensional measurements of a tumor were based on its longest diameter and the greatest perpendicular measurement from this diameter from CT scans.
RECIST criteria were not used in this Phase 2 clinical trial and instead the clinical investigator used the following definitions for tumor responses. A ‘‘complete response’’ was defined in this Phase 2 clinical trial as the clinically and radiologically documented complete disappearance of the principal target tumor or other target tumors, based on bidimensional measurement as determined by two radiological observations not less than four weeks apart. A ‘‘partial response’’ was defined in this Phase 2 clinical trial as a 50% or more decrease of the bidimensional measurements in the principal target tumor that had been measured as compared to baseline. ‘‘Progressive disease’’ was defined in this Phase 2 clinical trial as either: at least a 25% increase of the bidimensional measurements for tumors greater than four cm2 or at least a 50% increase of the bidimensional measurements for tumors less than four cm2 in the principal target tumor as compared to the nadir, which was defined in this Phase 2 clinical trial as the smallest radiologically determined tumor size achieved during the clinical trial. ‘‘Stable disease’’ was defined in this Phase 2 clinical trial as disease that meets neither the ‘‘partial response’’ nor the ‘‘progressive disease’’ criteria during or following active treatment and based on the sum of its bidimensional measurements, includes a less than 25% increase in tumor size for tumors greater than four cm2, or a less than 50% increase in tumor size for tumors less than four cm2. Tumor measurements that were radiographically confirmed showed that four of the eight evaluable subjects (50%) demonstrated ‘‘stable disease’’ for their principal tumor.
When radiographic best responses were evaluated at any two treatment time points, including baseline, 13 of the 14 evaluable subjects (92.9%) showed ‘‘stable disease’’ or partial response, with 10 of the 14 evaluable subjects (71.4%) showing a decrease in tumor size ranging from 4% to 85%. Measurements of the principal target tumors taken at baseline and at final visit showed that three of the eight evaluable subjects (37.5%) had decreases in principal tumor size ranging from 4% to 35%. Of five subjects with multiple tumors who achieved primary tumor responses, four of those subjects also achieved responses in subsequently injected non-primary tumors. These results suggest that Proxinium may be effective in the treatment of EpCAM-positive late-stage SCCHN.
This Phase 2 clinical trial was completed in August 2007.
Phase 3 clinical trial. The company initiated a randomized Phase 3 clinical trial of Proxinium in January 2006 at 75 sites globally. Total enrollment was planned for 292 subjects with late-stage SCCHN and the protocol included two periods: a Phase 2 lead-in period comprised of 30 subjects and a Phase 3 period comprised of 262 subjects. Each subject’s locally recurrent disease had to be refractory, which means that the subject must have progressed on or after receiving, or was unable to tolerate, at least one anti-cancer treatment regimen or had to have previously documented refusal of treatment for locally recurrent disease.
This Phase 3 clinical trial was conducted to compare the overall survival time associated with intratumorally injected Proxinium and safety and efficacy data of Proxinium in combination with BSC versus BSC alone, in subjects with locally recurrent disease who had received at least one anti-cancer treatment regimen for such locally recurrent disease. Secondary objectives were to compare the loco-regional response rate and duration of loco-regional response, the local progression-free survival, symptomatic benefit and safety profile for subjects from the Proxinium in combination with BSC arm and the BSC alone arm.
During the treatment phase of this Phase 3 clinical trial, all subjects were assessed weekly and treated according to institutional standards of BSC, which included treatment measures such as the use of pain medication, hydration, antiemetics and nutritional support, but did not include the use of radiotherapy (except as needed for the palliation of distant metastases) or any agent that may have had an impact on tumor response. Subjects who were randomized to the BSC alone arm were either seen in the clinic or had weekly assessments conducted by phone; provided that at least one in-person visit was conducted every four weeks. Subjects who were randomized to Proxinium in combination with the BSC arm were also to receive BSC, as well as a once weekly intratumoral injection of Proxinium at 700 µg per dose. Subjects in both arms of this Phase 3 clinical trial continued in the treatment phase until either complete resolution of all target tumors or radiographic tumor progression occurred. All subjects were then to remain in the follow-up phase until one of the following occurred: ![]() 12 months from the date that the last subject required for efficacy analyses had been randomized, died, withdrew or the company terminated the trial or (ii) termination of the trial for safety reasons due to DLTs.
12 months from the date that the last subject required for efficacy analyses had been randomized, died, withdrew or the company terminated the trial or (ii) termination of the trial for safety reasons due to DLTs.
The intention of the Phase 2 period of the clinical trial was evaluation of available data once the first 30 subjects reached the four week treatment time point. Of the first 30 subjects enrolled, 15 were randomized to each study arm. The Phase 2 lead in period was specifically designed for the assessment of safety, with an independent review of the safety data by the data safety monitoring board, or DSMB. Following the review by the DSMB, they recommended the continuation of enrollment and monitoring as mandated by the protocol since the available data indicated that intratumoral administration of Proxinium was generally well-tolerated by the subjects. The Phase 3 period began immediately after the conclusion of the Phase 2 period, with no pause in enrollment.
There were 62 subjects for which post-treatment tumor measurements were available, 36 subjects in the Proxinium in combination with the BSC arm and 26 subjects in the BSC alone arm. Responses in FIGURE 5 below represent the best percentage change in radiographically determined bidimensional measurement from baseline at any time point for the injected tumor. Of the Proxinium in combination with BSC arm, 19 of the 36 subjects (52.8%) showed tumor reduction with a median reduction in bidimensional tumor measurement of 48.3%. In contrast, only 10 of the 26 subjects (38.5%) of the BSC alone arm showed a median reduction in tumor size of 21.9%. With respect to the subjects for whom the best bidimensional percent change showed an increase in tumor size, the Proxinium in combination with BSC arm had 12 of the 36 subjects (33.3%) showing a median increase of 31.7%. The increase in tumor size was more pronounced in the BSC alone arm with 11 of the 26 subjects (42.3%) showing a median increase of 60.8%.
Targeted tumor responses in FIGURE 5 below were categorized as ‘‘complete,’’ ‘‘partial,’’ ‘‘stable,’’ or ‘‘progressive’’ disease. A ‘‘complete’’ response was defined as radiographically confirmed complete disappearance of disease, ‘‘partial’’ response as a 50% or more decrease of the sum of the product of the bidimensional measurements as compared to baseline, and ‘‘progressive’’ disease as at least a 25% increase in the sum of the products of the bidimensional measurements compared to the radiographic nadir, when the sum at baseline was greater than four cm2 or at least a 50% increase of the sum of the product(s) of the bidimensional measurements compared to the radiographic nadir, when the sum at baseline was less than or equal to four cm2. ‘‘Stable’’ disease was defined as the response when neither the complete response, partial response nor the progressive disease criteria were met.
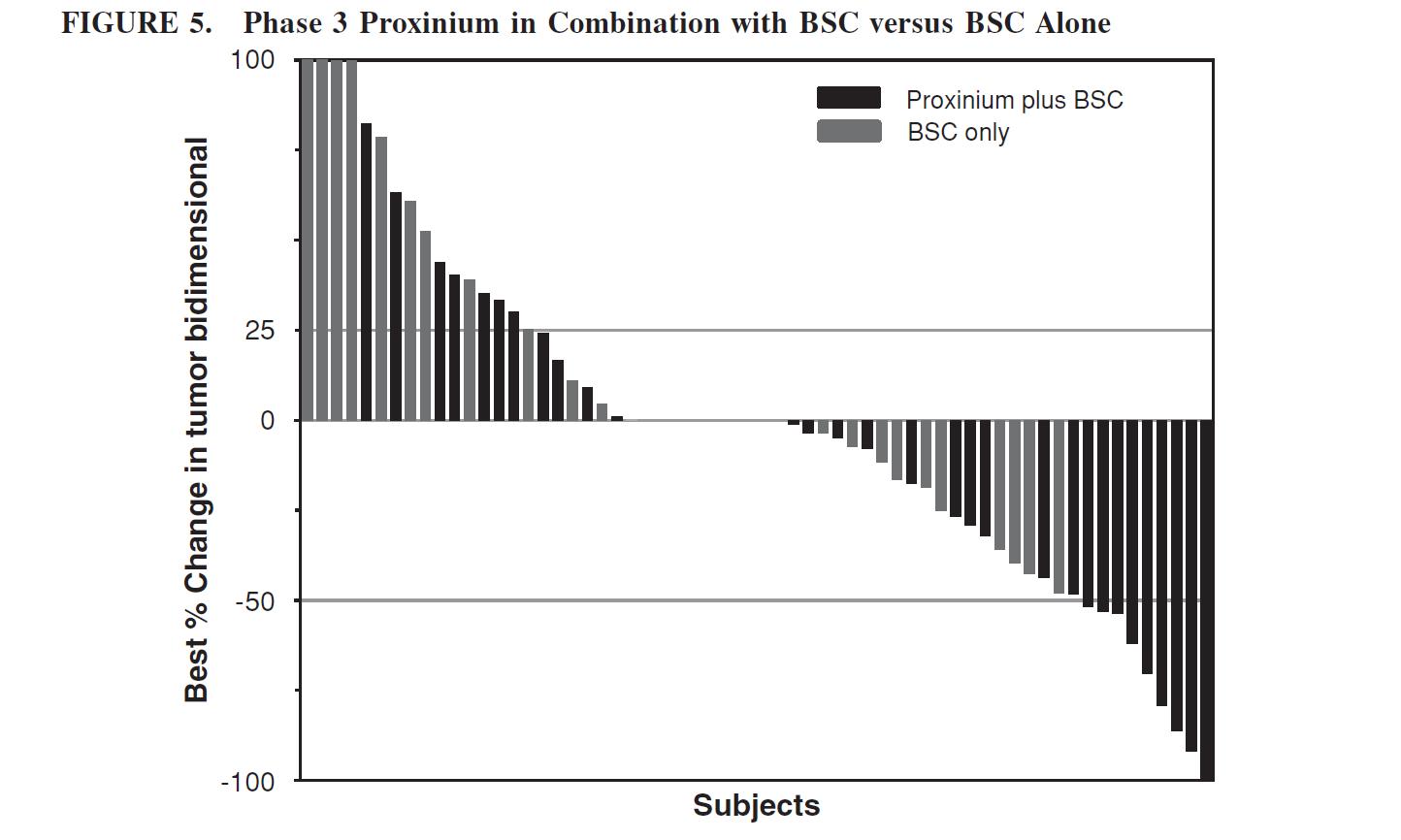
This Phase 3 clinical trial was terminated in April 2008 because of challenges relating to subject enrollment and retention for reasons specific to emerging markets, but not due to safety or efficacy concerns. Clinical trial subject enrollment and retention in emerging markets presents unique challenges compared to North America. With fewer established options for communicating with existing and new subjects, emerging market study centers have difficulty acquiring new subjects as well as maintaining consistent contact with existing subjects, making follow up very difficult. The company do not believe these issues will pose the same challenges in a North American clinical trial. In the United States, study centers have established multiple options to enroll and remain in communication with subjects.
At the time this Phase 3 clinical trial was terminated, 166 subjects had been randomized in the trial. Of these, 82 subjects were randomized to the Proxinium treatment in combination with the BSC arm and 84 subjects were randomized to the BSC alone arm (as discussed below).
Safety data. The company believe that its safety and anti-tumor activity data in its two Phase 1 clinical trials, its Phase 2 clinical trial, and its partially completed Phase 3 clinical trial support further development of Proxinium. There were no Grade 5 serious adverse events that were considered by the clinical investigator to be related to Proxinium. The serious adverse events that were reported in the clinical trials of Proxinium and were considered to be possibly, probably or definitely related to treatment consisted of abnormal tumor growth, anorexia, cancer pain, decrease in red blood cells, difficulty swallowing, elevated calcium levels, facial pain, fatigue, high blood sugar, influenza like illness, injection site pain, liver function abnormalities, low albumin level, low sodium concentration, nausea, rash, swelling, tumor hemorrhage and tumor necrosis.
Seven subjects died during the clinical trials of Proxinium, but none of the deaths were deemed to be related to Proxinium. Eleven subjects discontinued treatment due to liver function test abnormalities; however, the serum levels were transient and they eventually returned to baseline without any evidence of permanent liver damage. Four subjects withdrew from the clinical trials. Three of the four subjects withdrew at their request and one of the four subjects withdrew at the request of the investigator.
Proxinium Proposed Phase 1/2a Clinical Trial Plan
The company intend to initiate a Phase 1/2a clinical trial that will explore the potential of Proxinium in combination with a checkpoint inhibitor and are actively seeking partners for a combination program. The company anticipate that the Phase 1/2a clinical trial will explore the potential for Proxinium, due to its potential immunogenic effect, to enhance checkpoint inhibitors in combination therapy for the treatment of SCCHN.
Overall, the company believe that its efficacy and safety data support the continued clinical development of Proxinium to fulfill a significant unmet medical need in subjects with late-stage SCCHN.
Eleven Biotherapeutics has deferred further development of Proxinium in order to focus its efforts and its resources on its ongoing development of Vicinium. Eleven Biotherapeutics is also exploring collaborations for Proxinium.
Potential future indications
Based on the safety and efficacy data in its two Phase 1 clinical trials, its Phase 2 clinical trial and its previous partially completed Phase 3 clinical trial of Proxinium, the company also believe that there are several other potential applications for Proxinium that the company may elect to pursue, including colon, thyroid and prostate cancers.
VB6-845d
Overview
The company's lead systemically-administered product candidate, VB6-845d, is being developed as a treatment for multiple types of EpCAM-positive solid tumors. VB6-845d is a TPT consisting of an EpCAM targeting Fab genetically linked to deBouganin, which is administered by intravenous infusion. EpCAM is over-expressed on the cell surface of many solid tumors, including breast, colorectal, gastric, lung, ovarian and prostate. EpCAM overexpression has been shown to be involved in promoting malignant progression. In addition, EpCAM overexpression is associated with increased tumor grade, disease progression, increased proliferative phenotypes and diminished survival. EpCAM is also a cancer stem cell marker. A Phase 1 clinical trial conducted with VB6-845, the prior version of VB6-845d, revealed no clinically relevant immune response to the deBouganin payload, and five of seven subjects (71.4%) maintained stable disease (meaning no change in tumor size from baseline) after one completed cycle of treatment (four weeks) two subjects had decreases in target tumor size, and one subject who continued treatment through a third cycle (12 weeks) maintained stable disease. Interim safety data from its Phase 1 clinical trial was consistent with expectations for the study population of subjects with advanced solid tumors and the anticipated effects of targeted biological therapies containing immunogenic sequences.
Based upon the hypersensitivity reactions seen in its Phase 1 clinical trial conducted in Russia and in the country of Georgia, the company de-immunized the Fab portion of VB6-845 to create VB6-845d. In April 2016, the company submitted an IND to the FDA in preparation of initiating a Phase 1/2 clinical trial of VB6-845d in subjects with EpCAM-positive cancers in the United States. The IND was withdrawn in July 2016 after the company received initial feedback from the FDA indicating that they had identified hold and non-hold deficiencies that needed to be addressed. In December 2016, the company submitted a request for a pre-IND meeting to seek input on the manufacturing, nonclinical and clinical plans for VB6-845d prior to resubmitting an IND. In February 2017, the FDA provided guidance on its manufacturing and nonclinical plans for VB6-845d. Based on this guidance, Eleven Biotherapeutics is performing additional studies and the company plan on submitting an updated IND, once funding or a partner is secured for this program.
Overall, the company believe that its pre-clinical data and the interim Phase 1 clinical data support further clinical investigation of VB6-845d to explore whether it may fulfill the significant unmet medical need in the treatment of subjects with EpCAM-positive solid tumors. Specifically, the company believe that VB6-845d has potential to be a first-in-class TPT capable of providing clinical benefit in these difficult to treat patient populations.
Eleven Biotherapeutics is currently developing VB6-845d, a recombinant fusion protein consisting of an anti-EpCAM fragment fused to a deBouganin payload for the systemic treatment of advanced solid tumors. DeBouganin acts by inhibiting protein synthesis and helps circumvent multi-drug resistance mechanisms. Solid tumors form an abnormal and discrete tumor mass in the body that usually does not contain cysts or liquid areas.
The company believe that its TPTs utilizing its de-immunized deBouganin payload may be enhanced if combined with checkpoint inhibitors. The company believe that deBouganin’s potential effect on cancer cells could promote an immunogenic response that may enhance the action of checkpoint inhibitors.
Eleven Biotherapeutics has deferred further development of VB6-845d in order to focus its efforts and its resources on its ongoing development of Vicinium. Eleven Biotherapeutics is also exploring collaborations for VB6-845d.
The company own or exclusively license worldwide rights to its VB6-845d intellectual property portfolio that provides unextended patent term until at least 2025 and, if its pending composition of matter patent applications for VB6-845d are granted until at least 2036. See ‘‘Business-Intellectual Property’’ for additional details.
Clinical trials and pre-clinical studies
Pre-clinical studies. VB6-845, the prior version of VB6-845d, demonstrated strong binding and potent activity with small doses of the product candidate against numerous EpCAM-positive solid tumor cell lines, including those derived from tumors of the breast, cervix, colon, endometrium, gastric, lung and ovary, in in vitro pharmacology studies. In vitro and in vivo pre-clinical data have also demonstrated that VB6-845 preferentially binds to tumor cells expressing EpCAM and is effective in inhibiting tumor growth and increasing survival in mouse models. These xenograft studies demonstrated that VB6-845 was able to selectively affect EpCAM-expressing tumors without observed systemic toxicity.
Phase 1 clinical trial. The company received regulatory approval from the Federal Service on Surveillance in Healthcare and Social Development of the Russian Federation (Roszdravnadzor) to conduct a Phase 1 clinical trial in Russia in March 2007 and from Ministry of Labour Health and Social Affairs of Georgia in April 2007. The company initiated an open-label Phase 1 clinical trial of VB6-845 in May 2007 at five sites in Russia and one site in Georgia. The company enrolled 15 subjects with EpCAM-positive solid tumors, including breast, colorectal, kidney, non-small cell lung, ovary, pancreas and stomach cancers. All subjects enrolled in this Phase 1 clinical trial had tumors that tested positive for EpCAM as confirmed by its immunohistochemical test used during this Phase 1 clinical trial of VB6-845. The Phase 1 clinical trial was designed to define the maximum tolerated dose and to evaluate immunogenicity, safety. Secondary objectives included information on PK properties and assessing exploratory efficacy of VB6-845.
Subjects were treated over three dose cohorts in this Phase 1 clinical trial. VB6-845 was administered as a monotherapy intravenous infusion (for a period of over three hours), once weekly in four-week cycles, to subjects with EpCAM-positive advanced solid tumors. Subjects were evaluated at the end of each cycle. Three subjects at the first cohort dose level received 1.00 mg/kg, 10 subjects at the second cohort dose level received 2.00 mg/kg and two subjects at the third cohort dose level received 3.34 mg/kg. Following treatment of the subjects in the second cohort, the clinical reporting company reviewed the safety data and unanimously decided to escalate to the third cohort at a dose of 3.34 mg/kg. Out of the first group of five subjects in the second cohort, one DLT (an infusion-related reaction) was reported and confirmed by the clinical trial monitoring company. As the study was stopped early, no maximum tolerated dose was reached. VB6-845 was generally well-tolerated up to the third dose cohort (3.34 mg/kg).
One of its primary objectives in this Phase 1 clinical trial was to validate the extensive pre-clinical data supporting de-immunization of the deBouganin cytotoxic protein payload in humans. For a payload to be viable systemically it must be de-immunized to prevent rapid clearance by the immune system. Subject blood samples for the assessment of an anti-VB6-845 response were collected pre-infusion for the first cycle and every four weeks thereafter. An analysis of the antibody titers at each time point revealed a minimal immune response directed against the deBouganin moiety for only two subjects after eight weeks. Moreover, the titer of the immune response was just above the threshold of the assay (ranging between a titer of 1500 to 1800). These findings are consistent with significant de-immunization of the parent Bouganin cytotoxic protein payload via T-cell epitope depletion.
For all cohorts, exploratory efficacy data (CT and radiographic) were available for seven subjects who completed one full cycle (four weeks) of treatment. Five of the seven subjects showed stable disease, which means tumor measurement is unchanged relative to baseline, on CT scans one week after the completion of a fourth dose of VB6-845. Of the three subjects who continued to receive treatment past the first cycle, one subject continued to have stable disease at the completion of their second (eight weeks) and third (12 weeks) cycles. There was radiographic evidence of decreases in tumor size in two subjects with renal cell carcinoma and breast carcinoma. For one subject, six measurable target tumors in the lungs as well as a measurable target tumor in a pulmonary lymph node and pelvic mesentery showed decreases by CT scan ranging from 11% to 29% on a final visit relative to the baseline. Other non-target, non-measurable tumors appeared unchanged; although there was an appearance of disease progression as a potentially new brain tumor, which was inaccessible to treatment. In one other subject, CT scan results revealed decreases in four of the five measurable target tumors in the liver, with decreases in individual tumors ranging from 4% to 15%. Non-target, non-measurable tumors in lungs, liver and bones showed stable disease.
This Phase 1 clinical trial was terminated in April 2008. Subjects in this Phase 1 clinical trial exhibited little to no immune responses to the deBouganin payload, thereby demonstrating de-immunization of deBouganin. Subjects did, however, generate antibodies against the Fab molecule in the product candidate, specifically against mouse amino acid sequences that were left in the Fab to increase the thermal stability in this early version of the molecule. Taken together, the company believe this demonstrated that the subjects were fully immunocompetent, which means that they were capable of rejecting the deBouganin payload if their immune system recognized it as foreign. Furthermore, it is important to note that the deBouganin payload was presented to the subjects’ immune system as a fusion with the immunogenic Fab fragment. The ‘‘hapten-carrier effect’’ principal in immunology dictates that presenting a non-immunogenic ‘‘hapten’’ protein (deBouganin) to the immune system as a conjugate or fusion to an immunogenic ‘‘carrier’’ protein (Fab with mouse amino acid sequences) is an effective way to amplify the immunogenicity of the non-immunogenic protein. The company's observation of a lack of immunogenicity of deBouganin in this setting is further evidence of its de-immunization. Eleven Biotherapeutics has since engineered these mouse amino acid sequences out of VB6-845, which the company refer to as VB6-845d, and based upon a binding specificity pre-clinical study, VB6-845d retained biologic activity.

The chart below demonstrates why the company believe that Eleven Biotherapeutics has successfully de-immunized VB6-845 to create VB6-845d as shown by in a Phase 1 clinical trial that revealed no clinically relevant immune response to the deBouganin payload.
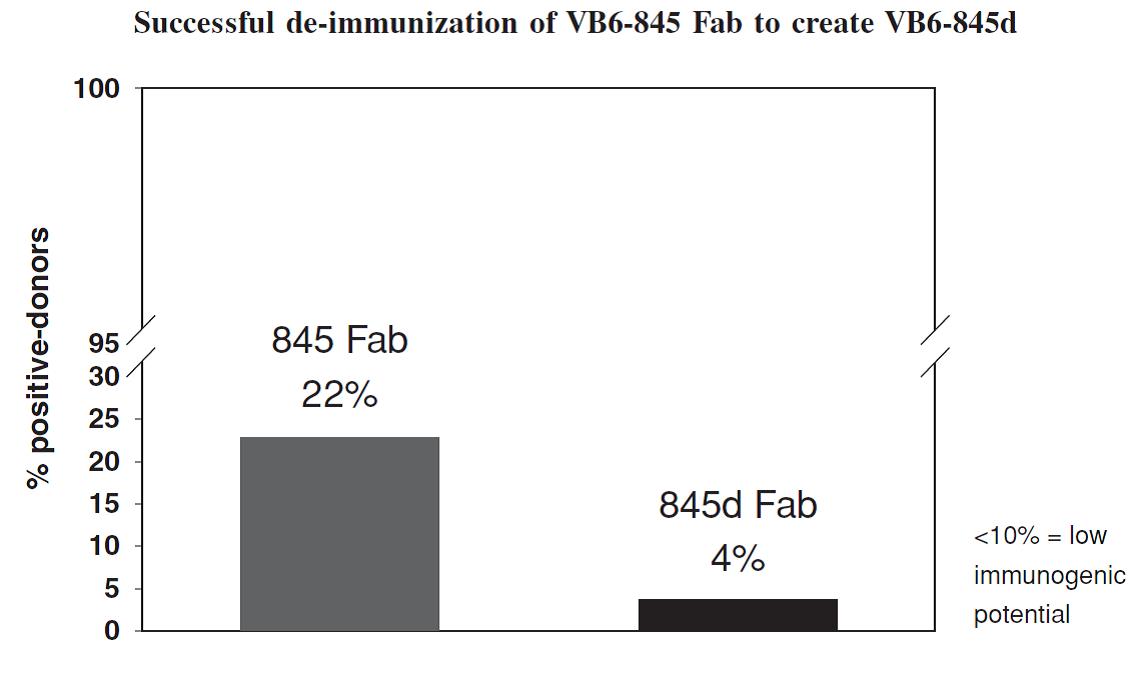
Safety data. The company believe that the interim safety data from the 15 subjects in its Phase 1 clinical trial support further development of VB6-845d. There were no Grade 5 serious adverse events that were considered by the clinical investigator to be related to VB6-845. The Grade 3 and Grade 4 serious adverse events that were reported in the Phase 1 clinical trial of VB6-845 and considered to be possibly, probably or definitely related to treatment, consisting of an infusion related reaction and an infusion site reaction that are consistent with the immunogenic nature of the Fab fragment. The subject’s condition improved and the event was considered to be resolved one day after onset without any further clinical concerns. The subject with the infusion related reaction was discontinued from the Phase 1 clinical trial in accordance with the protocol treatment stopping criteria defined for Grade 4 serious adverse events. The adverse event data reported for the subjects at the time the Phase 1 clinical trial was terminated was based upon interim data.
VB6-845d Phase1/2 clinical trial development plan
In April 2016, the company submitted an IND to the FDA in preparation of initiating a Phase 1/2 clinical trial of VB6-845d in subjects with EpCAM-positive cancers in the United States. The IND was withdrawn in July 2016 after the company received initial feedback from the FDA indicating that they had identified hold and non-hold deficiencies that needed to be addressed. In December 2016, the company submitted a request for a pre-IND meeting to seek input on the manufacturing, nonclinical and clinical plans for VB6-845d prior to resubmitting an IND. In February 2017, the FDA provided guidance on its manufacturing and nonclinical plans for VB6-845d. Based on this guidance, Eleven Biotherapeutics is performing additional studies and plan on submitting an updated IND.
Overall, the company believe that its pre-clinical data and the interim Phase 1 clinical data support further clinical investigation of VB6-845d to explore whether it may fulfill the significant unmet medical need in the treatment of subjects with EpCAM-positive solid tumors. The company also believe that the deBouganin payload in VB6-845d may enhance the action of checkpoint inhibitors as a result of the promotion of a local tumor immune response following the death of cancer cells.
EBI-031
License Agreement with Roche
On June 10, 2016, the company entered into the License Agreement with Roche. The License Agreement became effective on August 16, 2016, following stockholder approval. Under the License Agreement, the company granted Roche an exclusive, worldwide license, including the right to sublicense, to its patent rights and know-how related to its monoclonal antibody EBI-031 or any other IL-6 antagonist anti-IL-6 monoclonal antibody, to make, have made, use, have used, register, have registered, sell, have sold, offer for sale, import and export any product containing such an antibody or any companion diagnostic used to predict or monitor response to treatment with such a product, or collectively, Licensed Intellectual Property.
Pursuant to the terms of the License Agreement, Roche is required to continue developing EBI-031 and any other product made from the Licensed Intellectual Property that contains an IL-6 antagonist anti-IL-6 monoclonal antibody, or Licensed Product, at its cost.
Financial Terms
Roche paid an up-front license fee of $7.5 million upon effectiveness of the license under the License Agreement, and agreed to pay up to an additional $262.5 million upon the achievement of specified regulatory, development and commercial milestones with respect to up to two unrelated indications. Specifically, an aggregate amount of up to $197.5 million is payable to it for the achievement of specified milestones with respect to the first indication: consisting of $72.5 million in development milestones, $50.0 million in regulatory milestones and $75.0 million in commercialization milestones.
Roche paid the first development milestone of $22.5 million as a result of the IND application for EBI-031 becoming effective. Additional amounts of up to $65.0 million are payable upon the achievement of specified development and regulatory milestones in a second indication.
In addition, Eleven Biotherapeutics is entitled to receive royalty payments in accordance with a tiered royalty rate scale, with rates ranging from 7.5% to 15% for net sales of potential future products containing EBI-031 and up to 50% of these rates for net sales of potential future products containing other IL-6 compounds, with each of the royalties subject to reduction under certain circumstances and to the buy-out options of Roche further described below.
Buy-Out Options
The License Agreement provides for two “option periods” during which Roche may elect to make a one-time payment to it and, in turn, terminate its diligence, milestone and royalty payment obligations under the License Agreement. Specifically, ![]() Roche may exercise a buy-out option following the first dosing, or Initiation, in the first Phase 2 study for a Licensed Product until the day before Initiation of the first Phase 3 study for a Licensed Product, in which case Roche is required to pay it $135.0 million within 30 days after Roche’s exercise of such buy-out option and receipt of an invoice from it, or (ii) Roche may exercise a buy-out option following the day after Initiation of the first Phase 3 study for a Licensed Product until the day before the acceptance for review by the FDA or other regulatory authority of a biologics license application, or BLA, or similar application for marketing approval for a Licensed Product in either the United States or in the E.U. in which case Roche is required to pay it, within 30 days after Roche’s exercise of such buy-out option and receipt of an invoice from it, $265.0 million, which amount would be reduced to $220.0 million if none of its patent rights containing a composition of matter claim covering any compound or Licensed Product has issued in the E.U.
Roche may exercise a buy-out option following the first dosing, or Initiation, in the first Phase 2 study for a Licensed Product until the day before Initiation of the first Phase 3 study for a Licensed Product, in which case Roche is required to pay it $135.0 million within 30 days after Roche’s exercise of such buy-out option and receipt of an invoice from it, or (ii) Roche may exercise a buy-out option following the day after Initiation of the first Phase 3 study for a Licensed Product until the day before the acceptance for review by the FDA or other regulatory authority of a biologics license application, or BLA, or similar application for marketing approval for a Licensed Product in either the United States or in the E.U. in which case Roche is required to pay it, within 30 days after Roche’s exercise of such buy-out option and receipt of an invoice from it, $265.0 million, which amount would be reduced to $220.0 million if none of its patent rights containing a composition of matter claim covering any compound or Licensed Product has issued in the E.U.
Termination
Either the company or Roche may each terminate the License Agreement if the other party breaches any of its material obligations under the License Agreement and does not cure such breach within a specified cure period. Roche may terminate the License Agreement following effectiveness by providing advance written notice to it or by providing written notice if Eleven Biotherapeutics is debarred, disqualified, suspended, excluded, or otherwise declared ineligible from certain federal or state agencies or programs. The company may terminate the License Agreement if, prior to the first filing of a BLA for a Licensed Product, there is a period of 12 months where Roche is not conducting sufficient development activities with respect to the products made from the Licensed Intellectual Property.
Intellectual property
The company currently own or exclusively license approximately 17 families of patents and applications, which generally relate to its TPT-based product candidates and evolving its platform of targeting agents, cytotoxins (such as deBouganin) and linker technologies. As its product candidates evolve through clinical development, the company continue to monitor advancements and bolster patent coverage with the goal of attaining durable patent protection for at least 15 years from product launch. In addition, the company may prepare and file a number of additional applications around its platform technology, including its various targeting agents, cytotoxins, and linkers that, if issued, would expire in 2038 and beyond.
Product Candidates-Vicinium and Proxinium
The company exclusively license two families (70 patents and 3 applications) from the University of Zurich, or Zurich, which, among other things, include composition of matter claims directed to EpCAM antibody chimeras, EpCAM antibody chimera-cytotoxin conjugates, and their potential use in treating bladder and head and neck cancer. These families claim all or portions of Vicinium and Proxinium, as well as certain of their respective indications under clinical development. The first family includes composition of matter claims directed to the EpCAM antibody chimeras that are used in Vicinium and Proxinium. The first family consists of 21 patents in the United States, Canada, Europe and Japan, which expire in April 2020, subject to any applicable patent term adjustment or extension that may be available on a jurisdictional basis. The second family includes claims directed to the use of Vicinium and Proxinium in the treatment of bladder and head and neck cancer, respectively, and consists of 49 issued patents in the United States, Europe, Canada, China, Israel and Japan and pending applications in the United States, Canada, and Hong Kong. The expiry date of the patents in this family is April 2024, subject to any applicable patent term adjustment or extension that may be available on a jurisdictional basis.
In addition to the Zurich portfolio, the company own one issued U.S. patent with composition of matter claims directed to modified nucleic acid sequences that encode Vicinium and Proxinium and are potentially useful for high expression yield of Vicinium and Proxinium. The expiry date of this patent is in February 2029, subject to any applicable patent term extension that may be available on a jurisdictional basis. In addition, Eleven Biotherapeutics has filed and may prepare and file a number of additional patent applications with claims around composition of matter, manufacturing and purification methods, medical applications, and various uses of its various product candidates that, if issued, would expire in 2036 and beyond.
Bouganin and deBouganin family
The company exclusively license a family of patents and applications licensed from Merck KGaA, or Merck, which include claims directed to, among other things, modified de-immunized bouganin protein, EpCAM antibody-bouganin conjugates, and use claims directed to, among other things, methods of using the same to treat various diseases, including cancer. Claims in this family may cover, among other things, both the immunoconjugate, VB6-845d, and the de-immunized bouganin cytotoxins used in its product candidates. Currently the family consists of three issued patents in the United States, as well as 30 issued patents in Australia, Canada, China, Europe, Hong Kong, India, Israel, Japan, South Korea, Mexico, New Zealand, Russia and South Africa and one pending application in Brazil. The expiry date of this family is in March 2025, subject to any applicable patent term adjustment or extension that may be available on a jurisdictional basis. The company also exclusively license from Merck three additional families of patents and applications with, among other things, use claims directed to various de-immunization methodologies, which expire in May 2018, December 2018 and February 2022, subject to any applicable patent term adjustment or extension that may be available on a jurisdictional basis. In addition, Eleven Biotherapeutics has filed and may prepare and file a number of additional patent applications with, among other things, composition of matter and use claims around its various product candidates that, if issued, would expire in 2036 and beyond.
The company also exclusively license a family of patents directed to the unmodified bouganin cytotoxin from Protoden Technologies Inc., or Protoden, a company owned by Clairmark Investments Ltd., or Clairmark. See ‘‘See ‘‘Board Policies-Related Party Transactions’’ for additional details. The two United States patents expire in June 2018, subject to any applicable patent term adjustment or extension that may be available on a jurisdictional basis. The company do not currently view these patents and applications as significant to the development and commercialization, if approved, of its product candidates.
EBI-031 and its Legacy Product Candidates
As of March 30, 2018, the company owned the following families of patents and patent applications related to EBI-031 and its legacy product candidates, including EBI-005, or isunakinra. As of March 30, 2018, its patent portfolio includes the following patents and applications related to its legacy product candidates:
a United States, a New Zealand, Japan, Taiwan, China and a South Africa composition of matter patent covering isunakinra which expires in 2031;composition-of-matter patent applications covering isunakinra in Australia, Brazil, Canada, China, Europe, Hong Kong, India, Israel, Korea, Mexico and Russia, which, if granted, are expected to expire in 2031;patent applications covering the formulation of isunakinra filed in the United States, Australia, Canada, China, Europe, Hong Kong, Japan, New Zealand, Russia, and Singapore, which, if granted, are expected to expire in 2034;a patent application covering the formulation of isunakinra in a blow fill seal container filed in Taiwan, which if granted, is expected to expire in 2035;
Additional families of patent applications owned by Eleven include:
a provisional application directed to compositions and methods for increasing the retention of therapeutic agents in the eye which, if converted and granted, is expected to expire in 2038.a provisional application directed to compositions and methods for increasing the retention of anti-VEGF therapeutic agents in the eye which, if converted and granted, is expected to expire in 2038; anda provisional application directed to compositions and methods for increasing the retention of RGD therapeutic agents in the eye which, if converted and granted, is expected to expire in 2038
To the best of its knowledge based on correspondence received prior to April 21, 2017, the following families are owned by Eleven, and licensed to Roche pursuant to the License Agreement dated June 10, 2016:
patent applications covering the IL-6 antagonistic anti-IL6 monoclonal antibodies and active fragments thereof, including IL-6 antibody EBI-029, filed in the United States, Australia, Brazil, Canada, China, Europe, Hong Kong, India, Israel, Japan, Korea, Mexico, New Zealand, Russia, Singapore, and South Africa, and, if granted, are expected to expire in 2033;patent applications covering IL-6 antagonistic anti-IL6 monoclonal antibodies and active fragments thereof, including the IL-6 antibody EBI-031, having a pending PCT application and applications pending or to be filed in Algeria, Australia, Bahrain, Brazil, Canada, Chile, Colombia, Costa Rica, Egypt, Europe (to be filed), India, Israel, Korea, Malaysia, Mexico, Morocco, New Zealand, Oman, Philippines, Qatar, Russian Federation, Saudi Arabia, Singapore, South Africa, Thailand, Ukraine, United Arab Emirates, and Vietnam, and, if granted are expected to expire in 2035; anda PCT Application and an Argentine application each corresponding to a United States provisional application covering the IL-6 antibody EBI-031 formulation, which if granted, are expected to expire in 2036.
License Agreements
License agreement with The University of Zurich
Overview and exclusivity. Eleven Biotherapeutics has a license agreement with Zurich, which grants it exclusive rights, with the right to sublicense, to make, have made, use, and sell under certain patents primarily directed to its targeting agent, including EpCAM chimera, and related immunoconjugates and methods of use and manufacture of the same. These patents cover some key aspects of its product candidates Vicinium and Proxinium. Under the terms of the agreement, the company may be obligated to pay $750,000 in milestone payments, for the first product candidate that achieves applicable clinical development milestones. Based on current clinical status, the company anticipate that these milestones may be triggered by Vicinium’s clinical development pathway. As part of the consideration, the company will also be obligated to pay a 4% royalty on the net product sales for products covered by or manufactured using a method covered by a valid claim in the Zurich patent rights. Eleven Biotherapeutics has the right to reduce the obligation to pay royalties in a particular country expires upon the expiration or termination of the last of the Zurich patent rights that covers the manufacture, use or sale of a product. There is no obligation to pay royalties in a country if there is no valid claim that covers the product or a method of manufacturing the product. As of the date of this Annual Report on Form 10-K, aggregate license fees of $250,000 have been paid to Zurich by Viventia prior to its acquisition of Viventia. There were no payment made for the year ended December 31, 2017.
Patent rights. Eleven Biotherapeutics is responsible for the patent filing, prosecution and maintenance activities pertaining to the patent rights, at its sole expense, while Zurich is afforded reasonable opportunities to review and comment on such activities. If appropriate, the company shall apply for an extension of the term of any licensed patent where available, for example, in at least the United States, Europe and Japan. In the event of any substantial infringement of the patent rights, the company may request Zurich to take action to enforce the licensed patents against third parties. If the infringing activity is not abated within 90 days and Zurich has elected not to take legal action, the company may take legal action (in Zurich’s name, if necessary). Such action will be at its own expense and Zurich will have the opportunity to join at its own expense. Recoveries from any action shall generally belong to the party bringing the suit, but (a) in the event that the company bring the action and an acceptable settlement or monetary damages are awarded, then Zurich will be reimbursed for any amount that would have been due to Zurich if the products sold by the infringer actually had been sold by it, or (b) in the event a joint legal action is brought, then the parties shall share the expense and recoveries shall be shared in proportion to the share of expense paid by the respective party. Each party is required to cooperate with the other in litigation proceedings at the expense of the party bringing the action. Term and termination. The term of the agreement expires as of the expiration date of the last patent to expire within the Zurich patent rights. Eleven Biotherapeutics is currently projecting an expiration date for the U.S. licensed patents in 2024, subject to any applicable patent term adjustment or extension that may be available on a jurisdictional basis. Zurich has the right to terminate the agreement if the company breach any obligation of the agreement and fail to cure such breach within the applicable cure periods. Eleven Biotherapeutics has the right to terminate the agreement at any time and for any reason by giving 90 days written notice to Zurich.
License Agreement with Merck KGaA
Overview and exclusivity. In March 2004, the company entered into an exclusive license agreement with Biovation Limited, subsequently acquired by Merck, which was subsequently amended and restated in October 2015. Pursuant to the agreement, the company were granted an exclusive license, with the right to sublicense, under certain patents and technology relating to the de-immunization of its cytotoxin Bouganin for therapeutic and in vivo diagnostic purposes in humans. The de-immunized cytotoxin is known as deBouganin, and has been incorporated into its product candidate, VB6-845d. Eleven Biotherapeutics has the worldwide exclusive right, with the right to sublicense, under the licensed patents and technology to, among other things, make, have made, use or sell products incorporating deBouganin.
As of the date of this Annual Report on Form 10-K, aggregate license fees of $225,000 have been paid to Merck by Viventia prior to its acquisition of Viventia. There were no payments made for the year ended December 31, 2017. Under the agreement, the company may be obligated to pay certain clinical development and regulatory milestones for each ‘‘licensed product’’: (A) $2,000,000 upon the start of the first Phase 3 clinical trial for a licensed product; (B) $2,000,000 upon submission of the first BLA for a licensed product; (C) $2,000,000 upon the approval of the first BLA in certain countries for a licensed product and $1,000,000 upon each of the second and third approvals of a BLA in certain additional countries for the same licensed product (total of $4,000,000); and (D) $2,000,000 upon the approval of the second BLA in certain countries for a licensed product; and $1,000,000 upon each of the second and third approvals of the second BLA in certain additional countries for the same licensed product (total of $4,000,000). As part of the consideration, Eleven Biotherapeutics is obligated to pay a 1.5% royalty on the net product sales up to $150,000,000 and a 2% royalty on the net product sales above such amount.
Patent rights. Eleven Biotherapeutics has the first right to file, prosecute and maintain licensed patents relating to de-immunized plasmids and proteins, including, among other things, its deBouganin and Merck has the first right to file, prosecute and maintain any other licensed patents. Eleven Biotherapeutics has the first right, but not the obligation, to enforce the licensed patents against third parties for suspected infringement, and, after repayment of costs and expenses, any recoveries under such suit will be treated as net product sales and the company shall pay a royalty on the same. The company may not settle such patent infringement suit without the prior written consent of Merck, but such consent shall not be unreasonably delayed or withheld. If the company decline to enforce the licensed patents against third parties for suspected infringement, Merck may bring such a patent infringement suit and any recoveries will be retained by Merck.
Term and termination. The agreement expires on a country-by-country and product-by-product basis until the longer of ![]() the expiration of the last to expire patent within the licensed patent rights that covers a licensed product and (ii) 10 years from the first commercial sale of a licensed product in such country; provided that no royalty is payable for more than 15 years from the first commercial launch of a licensed product anywhere in the world. Either party has the right to terminate the agreement for breach of the agreement and if the other party fails to cure such breach within the applicable cure period. Eleven Biotherapeutics has the right to terminate the agreement by giving Merck six months prior written notice.
the expiration of the last to expire patent within the licensed patent rights that covers a licensed product and (ii) 10 years from the first commercial sale of a licensed product in such country; provided that no royalty is payable for more than 15 years from the first commercial launch of a licensed product anywhere in the world. Either party has the right to terminate the agreement for breach of the agreement and if the other party fails to cure such breach within the applicable cure period. Eleven Biotherapeutics has the right to terminate the agreement by giving Merck six months prior written notice.
Manufacturing
The company maintain an approximately 31,400 square foot manufacturing, laboratory, warehouse and office facility in Winnipeg, Manitoba, Canada. Eleven Biotherapeutics has three 15 liter fermentors, one 150 liter fermentor, one 500 liter fermentor and one 1,500 liter fermentor. The company's classified fermentation suite and post-production processing capabilities were dedicated to producing its pre-clinical study and clinical trial batches. In September 2017, the company completed the manufacturing of all Vicinium necessary for its ongoing Phase 3 registration trial in patients with NMIBC, and for its CRADA with the NCI. In conjunction with this achievement, Eleven Biotherapeutics is ending its large-scale manufacturing activities and redirecting resources toward completing its Phase 3 trial. In the event the company obtain approval from the FDA to market any of its product candidates, the company will need to outsource its commercial scale manufacturing to contract manufacturing organizations, or CMOs. Eleven Biotherapeutics is currently in the process of identifying a CMO and evaluating the manufacturing technology transfer process to that CMO.
Commercial Operations
The company do not currently have an organization structured for the sales, marketing and distribution of products. The company may rely on licensing and co-promotion agreements with strategic collaborators for the commercialization of its products in the United States and other territories. If the company choose to build a commercial infrastructure to support marketing in the United States, such commercial infrastructure could be expected to include a sales force supported by sales management, internal sales support, an internal marketing group and distribution support. To develop the appropriate commercial infrastructure internally, the company would have to invest financial and management resources, some of which would have to be deployed prior to the approval of any of its product candidates.
Competition
The pharmaceutical industry is highly competitive, subject to rapid and significant technological change and has a strong emphasis on developing proprietary products. While the company believe that its next generation TPT platform, knowledge, experience and scientific resources provide it with competitive advantages, the company face competition from both large and small pharmaceutical and biotechnology companies, academic institutions and other research organizations; specifically with companies, institutions and organizations that are actively researching and developing products that attach proprietary cell-killing payloads to antibodies for targeted delivery to cancer cells. The company's competitors include, but are not limited to:
NMIBC: Aadi, LLC (ABI-009), Altor Bioscience Corporation (ALT-801), Cold Genesys, Inc. (CG0070), Endo Pharmaceuticals Inc. (Valstar) (approved drug), FKD Therapies Oy (Instilidrin), Merck and other pharmaceutical companies (BCG) (approved drug), Eli Lilly and Company (Gemcitabine) and Telormedix SA (Vesimune);SCCHN: Bristol-Myers Squibb Company (nivolumab)(approved drug), Eli Lilly and Company, and Merck (Erbitux, pembrolizumab) (approved drugs);Multiple types of solid tumors: Amgen Inc. (Panitumumab) (approved drug), Bayer AG and Onyx Pharmaceuticals (Sorafenib) (approved drug), Bristol-Myers Squibb Company, Eli Lilly and Company, and Merck (Erbitux) (approved drug), F. Hoffmann-La Roche AG (Bevacizumab) (approved drug), Genentech, Inc. (Bevacizumab, Erlotinib and Trastuzumab) (approved drugs), Pfizer, Inc. (Sunitinib) and Trion Research GmbH (Removab); andIn addition to competition from alternative treatments, the company may also face competition from products that are biosimilar to, and possibly interchangeable with, its product candidates. Biosimilar products are expected to become available over the coming years. Even if its product candidates achieve marketing approval, they may be priced at a significant premium over competitive biosimilar products if any have been approved by then and insurers or other third party payors may encourage or even require the use of lower priced biosimilar products. Even if its treatments receive market authorization, they may not be listed on the formularies of payors (public or private insurers) or reimbursed. This may impact the uptake of the drug as a treatment option for patients and/or the price at which the drug can be sold at. Further, if the drug is reimbursed it may be at a narrower indication than the full scope of market authorization.
Many of its competitors have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical studies, conducting clinical trials, obtaining regulatory approval and marketing than the company do. These competitors are also active in seeking patent protection and licensing arrangements in anticipation of collecting royalties for use of technology that they have developed. Moreover, specialized biologics, biopharmaceutical and biotechnology companies may prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
The company's commercial opportunity could be substantially limited in the event that its competitors develop and commercialize products that are more effective, safer, less toxic, more convenient or cheaper than its comparable products. In geographies that are critical to its commercial success, competitors may also obtain regulatory approvals before it, resulting in its competitors building a strong market position in advance of its product’s entry. The company believe the factors determining the success of its programs will be the drug design, effectiveness against multi-drug resistance mechanisms, efficacy, safety, price and convenience of its product candidates.




