Aevi Genomic Medicine
Overview
Aevi Genomic (GNMX) is a clinical stage biopharmaceutical company with an emphasis on identifying the genetic drivers of disease and applying this understanding to the pursuit of differentiated novel therapies primarily for pediatric onset, life-altering diseases, including rare and orphan diseases. The company look to find treatments for genetically defined diseases for which there are limited therapeutic options currently available, with a primary focus on pediatric patients. This strategy begins with identifying and genetically validating a therapeutic target and using genomics to guide product development. The strategy also involves identifying and acquiring otherwise abandoned or overlooked drug candidates and matching targets and mechanisms of action to novel genetic discoveries.1
Aevi Genomic has partnered with the Center for Applied Genomics, or CAG, at The Children’s Hospital of Philadelphia, or CHOP, to implement a genomic medicine driven approach to drug development. Included in the assets at CAG is a fully automated biorepository containing specimens from more than 75,000 pediatric patients and 150,000 relatives of those patients. The sample is highly enriched for rare and orphan diseases and the large majority of patients have been genotyped. Their phenotypes are recorded in a modern electronic health record that is linked to the genomics database and biorepository. The patients in the database have consented to anonymized use of their data for research and follow up contact if needed.
CAG continues to discover important and novel genetic biomarkers by both genome-wide association studies and exome sequencing and analysis of affected individuals and their family members. Such markers not only identify patients with the disease but frequently point to the cause of the disease and suggest targets and feasible intervention strategies that include protein or peptide therapy, monoclonal antibodies, drugs or gene therapy. By working initially in pediatric populations of specific diseases, the company can minimize the confounding environmental factors seen in older patients. In addition, the availability of robust genetic biomarkers allows it to design trials that focus on a highly-enriched patient population that the company believe is more likely to respond to targeted therapies and further enhance the likelihood of clinical and regulatory success. The company believe this will allow it to implement clinical development programs that will lead to higher value medicines that can address critical needs in patients suffering from rare and orphan diseases.
Product Pipeline
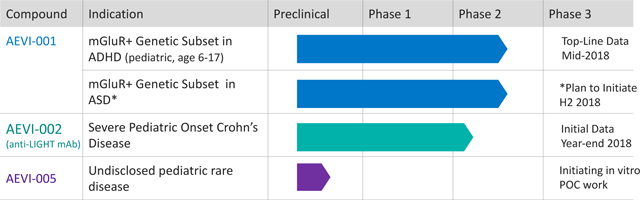
AEVI-001 (mGluR+ Genetic Subset ADHD)
The lead program from its genomic research collaboration with CHOP is the development candidate AEVI-001, an oral, non-stimulant glutamatergic neuromodulator. Through its acquisition of neuroFix, LLC, or neuroFix, in September 2015, the company acquired the rights to develop AEVI-001 (then known as NFC-1), as well as the rights to certain data derived from a clinical trial and other studies of AEVI-001.
The selection of AEVI-001 for development in the mGluR+ ADHD patients was the result of a rational search process conducted to specifically identify therapeutic candidates with a demonstrated ability to modulate glutamate signaling via the mGluR network. The role of glutamate in ADHD and other CNS disorders is supported by recent neuroimaging studies that suggest glutamate levels are abnormal in children with ADHD. These abnormalities appear to be concentrated in the anterior singular cortex region of the brain, as evidenced by volumetric and functional magnetic resonance imagery studies, as well as targeted studies of magnetic resonance spectroscopy. Additional supportive evidence for targeting glutamate modulation is provided by genetic studies that have identified mutations in glutamatergic genes that are enriched in children with ADHD.
ADHD Opportunity
Aevi Genomic is developing AEVI-001 to treat a sub-population of ADHD patients who have genetic mutations that disrupt the mGluR network, resulting in glutamate imbalance. ADHD is one of the most common childhood neurodevelopmental disorders of childhood. In the United States, the Center for Disease Control estimates that 6.4 million children 4-17 years of age (11%) have been diagnosed with ADHD. It is usually first diagnosed in childhood and often lasts into adulthood. Approximately 25% of ADHD patients are mGluR mutation positive, thereby representing approximately 1.5 million pediatric and 2.5 million adult patients in the United States. Based on pricing assumptions of currently available ADHD therapies, as well as established compliance and adherence rates, this equates to a potential $2 billion to $3 billion market opportunity for the drug.
ADHD is defined as a persistent pattern of inattention and/or hyperactivity-impulsivity that interferes with functioning or development. ADHD causes significant impairment in childhood and throughout the lifespan, as well as increased mortality and psychosocial adversity. There is no definitive management for ADHD; current management frequently includes a combination of educational support, behavioral interventions, and pharmacotherapy. Current standard of care is the stimulant class of medications including immediate- and extended-release methylphenidate and amphetamine; these products represent 90% of sales in the United States. In 2016, ADHD pharmaceutical product sales in the United States were approximately $11 billion, and grew at a compounded annual growth rate of approximately 2% from 2012 to 2016. However, while conferring great benefit for many individuals, currently available ADHD medications also have significant limitations including decreased appetite, weight loss, and insomnia.
Prevalence of mGluR Network Mutations
To examine the prevalence of mGluR network mutations in the broader pediatric and adolescent ADHD populations, the company conducted a large-scale non-interventional phenotype/genotype study at 32 sites across the United States. The study genotyped 1,876 ADHD patients aged 6-17 years, with 420 children and adolescents being mGluR+ (22.4%). A higher prevalence (75/292, 26%) was seen in patients aged 6-12 years than patients aged 13-17 years (344/1584, 21%). The data also showed that patients with the mGluR mutations had significantly higher prevalence of symptoms associated with inappropriate movements, disruptive behavior, and anger control.
mGluR Network Mutations Highly Predictive of ADHD
A study genotyped 3,445 ADHD patients from the CHOP Psychiatry and Behavioral Sciences Clinics to classify the prevalence of copy number variation mGluR+ mutations and the proportion of those patients who had already been diagnosed with ADHD. The research demonstrated the association between the excitatory glutamate neurotransmitter in the brain, mutations in the mGluR pathway, and ADHD in pediatric patients who possess these mutations. The study also clearly demonstrated the highly predictive capabilities of the genetic biomarker, as demonstrated by the fact that 98% of the patients with the identified mGluR network mutations had a positive diagnosis of ADHD (the study was conducted on a blinded basis). The company believe the genomic validation for AEVI-001 addresses a key inefficiency in the current ADHD diagnosis and treatment paradigm and may lead to improved safety and ultimately a personalized approach to treatment.
Development of AEVI-001 in mGluR+ Genetic Subset ADHD
AEVI-001 completed a Phase 2/3 trial (which the company refer to as the SAGA trial) in adolescent ADHD patients with specific mutations in their mGluR gene network, which the company refer to as mGluR+ ADHD, in the first quarter of 2017. Although AEVI-001 did not meet the primary endpoint of reduction on the ADHD rating scale (ADHD-RS) compared to placebo, in the SAGA trial, the drug did demonstrate statistically significant and clinically meaningful improvement compared to placebo in a pre-specified responder analysis of ADHD-RS improvement of 30% or more [ADHD-RS reduction of 17.6, p < .005]. In a second pre-specified responder analysis of Clinical Global Impression of Improvement scale (CGI-I), a key secondary endpoint, AEVI-001 demonstrated a statistically significant and clinically meaningful improvement compared to placebo [57% of patients treated with AEVI-001 achieved a score of much improved or very much improved compared to 33% on placebo, p=0.0155]. Additionally, the safety analysis demonstrated that AEVI-001 was well tolerated at all doses and the majority of adverse events were generally mild to moderate in severity. There were no serious adverse events.
Subsequent analysis of responder data from a subset of genomically identified patients in the SAGA trial identified nine genes (genetic subset) that appear to be predictive of clinically meaningful and statistically significant response on the ADHD-RS scales and CGI-I scales. These genes include certain glutamate metabotropic receptors and neurodevelopmental genes that are found in approximately 10% of pediatric ADHD patients.
One of the neurodevelopmental genes, contactin-4 (CNTN4), has been previously identified as being important in Autism Spectrum Disorder (ASD) representing approximately 5% of the overall pediatric ADHD patient population. The CNTN4 mutation phenotype is relatively severe, with an increased prevalence of emotional dysregulation, which includes issues related to anger control, risk taking, and inappropriate movements and sounds. All of the CNTN4 mutation positive (CNTN4+) patients on treatment (n=6, 100%) had clinically meaningful and statistically significant response to therapy with AEVI-001 [ADHD-RS reduction of 20.8, p=0.03].
Importantly, these results clarify a path forward for the continued development of AEVI-001 in ADHD, as well as in other potential neurodevelopmental disorders, including but not limited to ASD and Pediatric Generalized Anxiety Disorder. Aevi Genomic has initiated a Phase 2 trial in the mGluR mutation positive genetic subset ADHD (“mGluR+ Genetic Subset ADHD”) to confirm genetic responders to AEVI-001. Patient screening began in the third quarter of 2017 and data is expected by mid-2018.
In the United States, mGluR+ Genetic Subset ADHD represents approximately 10% of ADHD patients, estimated at 600,000 pediatric and 1.5 million adult. Based on pricing assumptions of currently available ADHD therapies, as well as established compliance and adherence rates, this equates to a potential $2 billion to $3 billion market opportunity for AEVI-001.
Diagnostic Development in ADHD
As part of its precision medicine strategy, Aevi is looking to develop and commercialize novel diagnostic tests to support therapies in development. For AEVI-001, Aevi is developing a stand-alone diagnostic to be used as an aid in the diagnosis of ADHD in patients aged 6-17, based on the discovery that mutations in the mGluR network are highly associated with ADHD. Aevi has engaged the US FDA on seeking a path to clearance for the diagnostic test. In addition to providing valuable information for the diagnosis of pediatric patients with mGluR+ ADHD, the diagnostic would support pre-identification of patients for future clinical trials in ADHD.
Previous Study of AEVI-001
The originator company for AEVI-001, Nippon Shinyaku, conducted research showing the ability of AEVI-001 to cross the blood-brain barrier and ameliorate cognitive impairment in animal behavioral models, at concentrations achievable in humans. AEVI-001 was shown to have a compelling pharmacokinetic and metabolic profile and to be a pan-selective activator and modulator of multiple mGluRs. Nippon Shinyaku studied AEVI-001 in vascular dementia, where approximately 1,000 adult patients were exposed to AEVI-001 for periods up to 12 months, in a development program that progressed to Phase 3. AEVI-001 was shown to be well tolerated with no treatment-emergent serious adverse events in this patient population, but was not effective for the treatment of vascular dementia.
The GREAT Study
A Phase Ib proof of concept trial (which the company refer to as the GREAT trial) of AEVI-001 in adolescent patients with ADHD was completed in 2015. The study enrolled 30 adolescents aged 12-17 with severe and genetically confirmed mGluR+ ADHD. Of the 30 enrolled patients, 17 had Tier 1 mGluR mutations, which are mutations in genes in the mGluR receptors or in genes that directly influence mGluR signaling. Seven patients had Tier 2 mutations, which are mutations in genes that encode proteins that influence mGluR. The remaining six patients had more distal Tier 3 mutations, which are mutations in genes that encode proteins that influence Tier 1 and Tier 2 genes.
Part 1 of the study measured safety and the pharmacokinetic profile of single ascending doses of 50-800mg of AEVI- 001. Part 2 of the study was single-blinded to patients and caregivers. Dosing was one week with placebo followed by four weeks of ascending doses from 50mg BID to 400mg BID of AEVI-001. The study used the Clinical Global Impression of Symptom Improvement (CGI-I) and the Vanderbilt Parent Rating Score (similar to the ADHD Rating Scale) to assess efficacy. Despite not being powered to show efficacy, the study demonstrated dose and duration-dependent improvements and response rates comparable to best-in-class ADHD therapies.
The treatment effect was more robust over time and at higher doses. In all patients, AEVI-001 showed weekly improvements in mean CGI-I from 3.79 during week 1 on placebo (baseline), 3.13 during week 2 (50mg BID), 2.79 during week 3 (100mg BID), 2.79 during week 4 (200mg BID) and 2.21 during week 5 (400mg BID). In all patients, AEVI-001 likewise showed weekly improvements in mean Vanderbilt scores from 29.1 during week 1 on placebo (baseline), 26.4 during week 2 (50mg BID), 24.0 during week 3 (100mg BID), 23.3 during week 4 (200mg BID) and 22.5 during week 5 (400mg BID).
The GREAT study also confirmed the previously observed pharmacokinetic profile of AEVI-001, showing the therapy to be well tolerated with no treatment-related SAEs. Following the conclusion of the study, a majority of patients enrolled in an open label long-term safety study. Full data from the study was presented at the American Academy of Child and Adolescent Psychiatry meeting in October 2015.
Development of AEVI-001 in 22q Deletion Syndrome (22q DS)
The company completed work on a signal-finding trial for the treatment of the psychiatric symptoms of 22q Deletion Syndrome (22q DS) in 2017. 22q DS is an orphan, severe autism spectrum disorder with significant co-morbidities. The disease has a prevalence of between 1:2000-1:4000, roughly equivalent with the more recognized Down’s Syndrome. Enrolling patients into the signal-finding study was difficult, with only two patients enrolled by the time the study ended. Due to the limited enrollment, it was not feasible to meaningfully interpret the resulting data, and the program was terminated.
Future Development of AEVI-001 in ASD
Aevi Genomic is exploring a development opportunity for AEVI-001 for the treatment of mGluR+ patients with ASD to better define the patient phenotype and intend to initiate work on a proof-of-concept study to begin in the second half of 2018. In 2012, 1 in 68 children were diagnosed with ASD in the United States, increasing from 1 in 150 in 2000. There is a high unmet need for pharmaceutical treatments for ASD as currently approved medications are indicated only for the symptoms of irritability in ASD patients. There are currently limited pharmacotherapy options available to treat ASD.
AEVI-002 (Anti-LIGHT Monoclonal Antibody)
The second program arising out of its genomic research collaboration with CHOP is the development candidate AEVI-002, a first-in-class anti-LIGHT monoclonal antibody, or the Antibody, being developed for use in Pediatric Onset Crohn’s disease. Pediatric Onset Crohn’s disease has a more aggressive phenotype at younger ages. The genomic rationale for the use of anti-LIGHT antibody in Crohn’s disease was validated by CAG research showing the association to a loss of function mutation in decoy receptor 3 (DcR3).
In June 2016, the company entered into a Clinical Development and Option Agreement, or the Development and Option Agreement, with Kyowa Hakko Kirin Co., Ltd., or KHK, pursuant to which the company acquired certain rights with respect to the development and potential commercialization of the Antibody. Under the Development and Option Agreement, the company received an exclusive option for exclusive rights to develop products containing the Antibody, or an Antibody Licensed Product, exclusive rights to commercialize Antibody Licensed Product in various countries and to conduct various development activities with respect to the Antibody Licensed Product, including the conduct of a signal finding study testing the Antibody in Severe Pediatric Onset Inflammatory Bowel Disease, or the Study. The terms of the Development and Option Agreement with KHK are more fully described under the section entitled “Licenses.”
An 8-week Phase Ib proof-of-concept study has been initiated at CHOP with the goal of enrolling up to 12 patients with a Pediatric Onset Crohn’s disease diagnosis with most patients being refractory to treatment with TNF-α inhibitors, with or without a DcR3 mutation. The endpoints of the trial will include endoscopic evaluation, Crohn’s Disease Activity Index ratings and safety. Initial data from the proof-of-concept study is expected by year-end 2018, at which point the company will make a determination on its option to license exclusive rights to the Antibody for further development. Active recruitment for the trial is underway, although the identification and recruitment of patients into the proof-of-concept study has been extremely challenging, and to date no patients have been enrolled. The ability to produce initial data by year-end 2018 is highly dependent on timely recruiting; thus, continued difficulties in recruitment could cause a delay in the delivery of initial data for the program. In an effort to address the recruitment challenges, Aevi Genomic is currently initiating three additional trial sites for the program.
Business Strategy
The company's goal is to translate key scientific insights relating to underlying genomic drivers of disease into the development of effective and highly selective therapeutics. To execute its strategy, the company intend to:
- Advance its lead product candidate AEVI-001 through clinical development. AEVI-001, a first-in-class non-stimulant mGluR modulator, is being developed for the treatment of mGluR+ Genetic Subset ADHD. AEVI-001 is currently being studied in a Phase 2 trial to confirm genetic responders to AEVI-001. Patient screening began in the third quarter of 2017 and data is expected by mid-2018.
- Pursue development of AEVI-001 for various other diseases where its genomic insights suggest it may be an effective therapy. In addition to mGluR+ Genetic Subset ADHD, the company intend to develop AEVI-001 for the treatment of certain genetically defined patient subsets with other neurological and neuropsychological indications, including but not limited to ASD.
- Advance its second clinical candidate AEVI-002 through clinical development. The second program arising out of its genomic research collaboration with CHOP is the development candidate AEVI-002, a first-in-class anti-LIGHT monoclonal antibody being developed for use in Pediatric Onset Crohn’s disease. An 8-week signal finding study at CHOP has been initiated and will enroll up to 12 patients with the DcR3 mutation and a Pediatric Onset Crohn’s disease diagnosis, with most subjects being refractory to treatment with TNF-α inhibitors. Initial data from the proof-of-concept study is expected by year-end 2018, at which point the company will make a determination on its option to license exclusive rights to the antibody for further development.
- Leverage its strategic collaborations to continue to implement a genomic medicine driven approach to drug development. The company's strategy is to work closely with its collaborators at CAG to identify populations of need with well-characterized, novel, genetically-defined targets. The company then designate an actionable therapeutic development approach based upon the target and the biology and human pathophysiology of the relevant disease and likely clinical and regulatory pathways. The collaboration affords it with unique and proprietary insight into these diseases and allows it to better select therapeutic approaches.
- Work with experienced third parties in the field of diagnostics. Because the company often target genetic alterations that are detectable, companion diagnostics can be developed to identify these alterations. Once Aevi Genomic has identified a target, the company will initially use existing diagnostic tools to identify patient subsets that the company believe will derive increased benefit from its product candidates. As the company advance its targets clinically and determine the most important screening criteria, the company will develop companion diagnostics as appropriate, with the help of technology partners, to identify patients and support registration and marketing of its product candidates.
- Opportunistically in-license and acquire novel therapies for the treatment of rare and orphan disease. The company plan to leverage its clinical drug development expertise and its relationships in the rare and orphan diseases community to identify and in-license or acquire additional product candidates that the company believe have the potential to become novel treatments for diseases with significant unmet medical needs.
- Potentially seek strategic collaborative relationships while maintaining flexibility in commercializing and maximizing the value of its development programs. The company plan to develop and seek regulatory approval for multiple product candidates in its development pipeline. While the company may develop these products independently, the company still may enter into strategic relationships with biotechnology or pharmaceutical companies to realize the full value of these products.
Intellectual Property
The company's goals are to obtain, maintain, and enforce patent and trademark protection for its products, processes, methods, and other proprietary technologies, including the platform collaboration with CHOP and to preserve its trade secrets both in the United States and elsewhere in the world. The company's policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for its products, processes and methods that arise from its genomics platform collaboration with CHOP through a combination of contractual arrangements, trade secrets, patents, and trademarks both in the United States and abroad.
The company's ability to compete depends on its ability to maintain and enforce its intellectual property rights and operating without infringing the intellectual property of others and its ability to enforce its licenses. The company's business could be materially harmed, and the company could be subject to liabilities, because of lawsuits brought by others against it or its licensors and licensees. The company will be able to protect its technology from unauthorized use by third parties only to the extent it is covered by valid and enforceable patents or is effectively maintained as trade secrets. Patents and other proprietary rights are an essential and material element of its business. Applications for patents and other intellectual property rights capable of being registered have been, and will be, filed in certain key jurisdictions. As the company identify additional rare and orphan disease targets, the company will seek protection for the related intellectual property rights in the United States and other relevant jurisdictions. There can be no assurance that the pending applications will result in patents ultimately being issued.
The company's patent portfolio for AEVI-001 and AEVI-002 consists of licensed patents and patent applications. The applicable licenses are discussed below.
The company also depend upon the skills, knowledge and experience of its scientific and technical personnel, as well as that of its advisors, consultants and other contractors, none of which is patentable. To help protect its proprietary knowledge and experience that is not patentable, and for inventions for which patents may be difficult to enforce, the company rely on trade secret protection and confidentiality agreements with its employees, consultants, vendors, collaborators, advisors, customers and other third parties to protect its interests. To this end, the company require all employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to it of the ideas, developments, discoveries and inventions important to its business. The company also require confidentiality or material transfer agreements from third parties that receive its confidential data or materials. The company intend to continue to take all appropriate steps to protect its intellectual property, including maintaining an active program for patent protection for novel elements in the development of its products and technology.
Licenses
neuroFix License
Immediately prior to and in connection with its acquisition of neuroFix in September 2015, neuroFix entered into a license agreement with CHOP, pursuant to which CHOP licensed to neuroFix certain technology owned and controlled by CHOP related to ADHD and certain other neurological and neuropsychological indications. Pursuant to this license agreement, CHOP licensed to neuroFix (coupled with a right to sublicense) certain patent rights and compound know-how on an exclusive, worldwide, royalty-bearing right and license basis, and certain CHOP know-how (other than compound know-how) on a non-exclusive, worldwide, royalty-bearing right and license basis. CHOP also granted to neuroFix an exclusive option during the term of the license agreement to negotiate an exclusive license to certain future CHOP intellectual property.
Pursuant to this license agreement, CHOP retained rights to the licensed patent rights and know-how to conduct teaching, educational, research and patient care activities itself and to conduct collaborations with certain not-for-profit, governmental, educational or non-commercial third parties and for purposes outside of the field of the license. Under the license agreement, neuroFix granted to CHOP a non-exclusive, worldwide, fully paid-up, royalty-free license under all intellectual property rights controlled by neuroFix to make and use certain products for education and non-commercial research purposes.
In addition to neuroFix having issued equity to CHOP in partial consideration for the rights granted under the license agreement (which equity was issued immediately prior to, and subsequently purchased by it in, to the acquisition), CHOP is eligible for certain milestone and royalty payments under the license agreement as further described below:
- up to $1.5 million in regulatory and sales milestone payments in connection with each FDA-approved indication obtained by neuroFix utilizing intellectual property licensed under the license agreement;
- royalty payments equal to a percentage of certain product sales by neuroFix using a fluctuating rate in the low single digits (adjusted downward to the extent third party royalty payments exceed a certain percentage in a given calendar quarter);
- annual maintenance fees of equal to or less than $100,000 depending on the year; and
- a certain percentage (ranging from mid-single digits to the mid-teens depending on if other rights of neuroFix are also licensed to the sublicensee at the same time) of all sublicensee income (except any amounts attributable to sublicensed sales by a certain party in Japan).
The license agreement will terminate, with respect to each product and each territory covered by the license agreement, upon the later of ![]() the expiration of certain CHOP patent rights and (ii) January 1, 2025, at which time the license rights granted to neuroFix become perpetual, irrevocable, fully paid-up and royalty-free. The license agreement could also be subject to termination by CHOP if neuroFix is not achieving certain specified development plans and diligence events and is not undertaking commercially reasonable efforts to achieve such events.
the expiration of certain CHOP patent rights and (ii) January 1, 2025, at which time the license rights granted to neuroFix become perpetual, irrevocable, fully paid-up and royalty-free. The license agreement could also be subject to termination by CHOP if neuroFix is not achieving certain specified development plans and diligence events and is not undertaking commercially reasonable efforts to achieve such events.
CHOP License Agreement and Sponsored Research Agreement
In November 2014, the company entered into a license agreement, or the License Agreement, and a sponsored research agreement, or the Research Agreement, each with CHOP. Under the terms of the License Agreement, CHOP granted it ![]() an exclusive, sublicensable license to use certain patent rights covering potential diagnostic and therapeutic targets, (ii) an exclusive, non-sublicensable license to use certain biospecimen and phenotypic data collected from patients with rare and orphan diseases and their family members, or the Biobank, (iii) a non-exclusive, sublicensable license to use certain know-how related to such patent rights, biospecimen and phenotypic data, (iv) a non-exclusive and non-sublicensable license to use certain biospecimen and phenotypic data collected from patients with non-rare and orphan diseases, and (v) an exclusive option to negotiate licenses to commercialize certain inventions that may be created in the future that target rare and orphan diseases. In consideration of the licenses and option granted under the License Agreement, the company agreed to pay to CHOP a license issuance fee of $500,000, certain maintenance fees, certain milestone payments, low single-digit royalties on net sales of all licensed products and a percentage of amounts received from sublicensing activities. In February 2017, the company amended the License Agreement. The amendment allows it to extend the period of its exclusive commercial access to the Biobank for rolling two year periods. The cost of each extension is $125,000 per year.
an exclusive, sublicensable license to use certain patent rights covering potential diagnostic and therapeutic targets, (ii) an exclusive, non-sublicensable license to use certain biospecimen and phenotypic data collected from patients with rare and orphan diseases and their family members, or the Biobank, (iii) a non-exclusive, sublicensable license to use certain know-how related to such patent rights, biospecimen and phenotypic data, (iv) a non-exclusive and non-sublicensable license to use certain biospecimen and phenotypic data collected from patients with non-rare and orphan diseases, and (v) an exclusive option to negotiate licenses to commercialize certain inventions that may be created in the future that target rare and orphan diseases. In consideration of the licenses and option granted under the License Agreement, the company agreed to pay to CHOP a license issuance fee of $500,000, certain maintenance fees, certain milestone payments, low single-digit royalties on net sales of all licensed products and a percentage of amounts received from sublicensing activities. In February 2017, the company amended the License Agreement. The amendment allows it to extend the period of its exclusive commercial access to the Biobank for rolling two year periods. The cost of each extension is $125,000 per year.
Under the terms of the Research Agreement, the company agreed to sponsor research at CHOP with respect to the recruitment and genetic analysis of patients with rare and/or orphan diseases to accelerate discovery of diagnostic and therapeutic targets. In February 2017, the company amended the License Agreement. The amendment allows it to extend the period of its exclusive commercial access to the Biobank for rolling two year periods. The cost of each extension is $125,000 per year. In June 2017, the company entered into an amendment to the Research Agreement, which extended the Research Agreement through June 30, 2019, for which payments totaling $5.94 million will be due in 2018 and $2.38 million will be due in 2019.
The License Agreement would terminate upon the expiration date of the last-to-expire royalty term under the License Agreement, however ![]() CHOP may terminate the License Agreement upon an uncured default by it or the failure by it to meet certain development and/or commercialization milestones under the License Agreement or if the company become insolvent or enter into bankruptcy proceedings, and (ii) the company may terminate the License at any time with six months prior written notice to CHOP.
CHOP may terminate the License Agreement upon an uncured default by it or the failure by it to meet certain development and/or commercialization milestones under the License Agreement or if the company become insolvent or enter into bankruptcy proceedings, and (ii) the company may terminate the License at any time with six months prior written notice to CHOP.
Development and Option Agreement, with Kyowa Hakko Kirin Co., Ltd. (KHK)
In June 2016, the company entered into the Development and Option Agreement with KHK pursuant to which the company acquired certain rights with respect to the development and potential commercialization of the Antibody. If the company exercise its option under the Development and Option Agreement, KHK has 60 days to select one of two development and commercialization structures as follows:
PLAN A: Co-Development/Co-Commercialization Arrangement
If KHK selects the co-development/co-commercialization arrangement (Plan A), the company will have the exclusive right to develop, manufacture and commercialize the Antibody Licensed Products in the treatment, prevention, and diagnosis of specified pediatric onset rare and orphan inflammatory diseases (including severe pediatric onset inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, or IBD) and other specified pediatric onset rare and orphan auto-immune diseases, or collectively, the Field, in the United States and Canada. The company will also be responsible for development and regulatory approval of the first Antibody Licensed Product in the European Union and then transferring such regulatory approval to KHK or its designee. The company will be responsible for the manufacture of the Antibody Licensed Products for use by the parties in clinical trials as well as for commercialization in their respective fields and/or territories, with KHK purchasing the Antibody Licensed Products from it.
The company will be required to pay KHK an initial license fee in the low single-digit millions of dollars upon the co-development/co-commercialization arrangement becoming effective. The company may pay KHK up to an additional $18 million upon the achievement of certain regulatory milestones related to the Antibody Licensed Products. The parties will share the anticipated costs of development of the first Antibody Licensed Product in the Field in the United States, Canada and the European Union with it being responsible for any costs in excess of an agreed cap. The parties will split profits from its sales of Antibody Licensed Products in the United States and Canada equally. KHK will pay it low double-digit royalties for sales of Antibody Licensed Products outside the United States and Canada and outside the Field in the United States and Canada.
PLAN B: Licensing Arrangement
If KHK selects the licensing arrangement (Plan B), the company will have the exclusive right to develop, manufacture and commercialize the Antibody Licensed Products in the Field in the United States, Canada and the European Union. The company will be responsible for the manufacture of the Antibody Licensed Products for use by the parties in clinical trials as well as for commercialization in their respective fields and/or territories.
The company will be required to pay KHK an initial license fee in the low single-digit millions of dollars upon the licensing arrangement becoming effective. The company may pay KHK up to an additional $28 million upon the achievement of certain regulatory milestones related to the Antibody Licensed Products. The parties will split profits from its sales of Antibody Licensed Products in the United States, Canada and the European Union with it being entitled to approximately 74% of such profits and KHK being entitled to approximately 26% of such profits. KHK will pay it low double-digit royalties for sales of Antibody Licensed Products outside the United States, Canada and the European Union and outside the Field in the United States, Canada and the European Union. The company will be responsible for costs of development of Licensed Products in the United States, Canada and the European Union. KHK will have the right to purchase the Antibody Licensed Products from it.
Properties
The company's principal executive offices are located at 435 Devon Park Drive, Suite 715, Wayne, Pennsylvania 19087. The company believe that this facility is adequate to meet its current needs. The company believe that if additional or alternative space is needed in the future, such space will be available on commercially reasonable terms as necessary.




