Anthera Pharmaceuticals
Overview
Anthera Pharmaceuticals, Inc. (ANTH) is a biopharmaceutical company focused on advancing the development and commercialization of innovative medicines that benefit patients with unmet medical needs. The company currently have two compounds in development, Sollpura and blisibimod. The company licensed Sollpura from Eli Lilly & Co (“Eli Lilly”) in July 2014. Sollpura is a novel non-porcine investigational Pancreatic Enzyme Replacement Therapy (“PERT”) intended for the treatment of patients with Exocrine Pancreatic Insufficiency (“EPI”), often seen in patients with cystic fibrosis and other conditions. The company licensed blisibimod from Amgen, Inc. (“Amgen”) in December 2007. Blisibimod targets B-cell activating factor, or BAFF, which has been shown to be elevated in a variety of B-cell mediated autoimmune diseases, including Immunoglobulin A nephropathy, or IgA nephropathy.
Sollpura
The exocrine pancreas is responsible for synthesis and secretion of digestive enzymes, including lipase, protease, and amylase. In addition, the pancreas secretes bicarbonate into the duodenum to neutralize the very high acidity of stomach contents. EPI occurs when diseases such as cystic fibrosis (“CF”) and chronic pancreatitis (“CP”) impede or destroy the exocrine function of the pancreas. A reduction in, or absence of, the normally secreted pancreatic digestive enzymes causes lipids, proteins, and carbohydrates to enter the distal gastrointestinal (“GI”) tract in un-absorbable forms, leading to GI pain and distention, mal-digestion, and steatorrhea. Without appropriate therapy, patients with EPI may experience malnutrition, poor growth, weight loss, reduced quality of life, and, in severe cases, increased morbidity and early death.
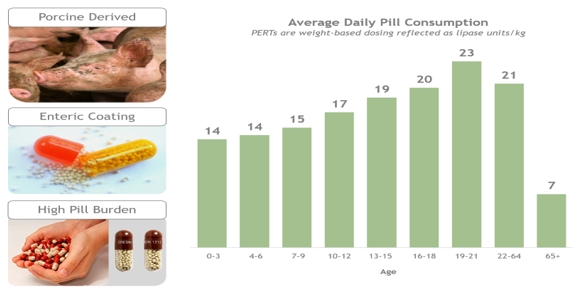
PERT is currently the mainstay of treatment for nutrient malabsorption in patients with digestive enzyme deficiencies known as EPI and orally delivered porcine PERTs have been available for many years for its treatment. As the porcine-derived proteins contained in the PERTs pass through the low pH environment of the stomach, they are protected by the enteric coating until they reach a pH of approximately 5.6 or greater in the duodenum and then are rapidly released. Due to the bulk imposed by the enteric coating, most patients have to take 10-20 capsules per day in order to maintain their nutritional requirement. Patient-to-patient differences in the acidity of the upper intestine makes dissolution of enterically-coated products variable, and gives rise to alterations in the rate and extent to which enzymes are released from these products. Poor stability and variability in terms of potency and pharmaceutical properties have also been identified as important factors contributing to a poor response of some patients to PERTs.
Current PERTs are Suboptimal
Sollpura is a novel, non-porcine PERT that contains three biotechnology-derived digestive enzymes: a lipase, a protease and an amylase. Through enzyme cross-linking, the lipase enzyme in Sollpura is more stable than the porcine-derived lipase in the low pH environment of the stomach and therefore Sollpura does not have an enteric polymer coating. Furthermore, since the three enzymes in Sollpura are biotechnology-derived, Sollpura does not contain porcine proteins or purines that may be associated with a risk of viral transmission or allergic reaction to proteins of porcine origin. The individual enzyme components of Sollpura are formulated at a fixed ratio of lipase, protease, and amylase. The Sollpura enzyme dose ratio was selected from nonclinical efficacy studies conducted using a canine model of pancreatic insufficiency which demonstrated that the lipase enzyme in Sollpura was efficacious when administered at >500 units/kg per meal, and the protease doses >1000 units/kg per meal. The suitability of the enzyme ratio in Sollpura is further supported by the observation that Sollpura and Creon were equi-effective in pigs with surgically-induced pancreatic insufficiency.
Sollpura – a Potentially Transformative Therapy for EIP
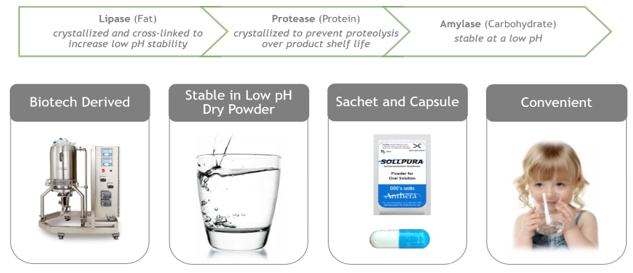
The company believe Sollpura has the potential to become the first soluble, stable and non-porcine derived enzyme product and offer a novel solution to patients who are unable to maintain appropriate nutritional health. Sollpura’s chemical characteristics, unlike currently available PERTs, make it ideal for powder formulation as either a capsule, or sachet of powder for oral solution which can be conveniently administered in solution in a small volume of water.
The company's Phase 3 Development of Sollpura in EPI
The company initiated a Phase 3 study of Sollpura ("RESULT") in patients with EPI due to cystic fibrosis in May 2017. The RESULT study is a randomized, open-label, assessor-blind, non-inferiority, active-comparator study evaluating the non-inferiority of Sollpura with respect to Coefficient of Fat Absorption (“CFA”) compared to a commercially available PERT in a population of porcine-derived PERT responders. The RESULT study’s design is modified from a previous Phase 3 study’s (“SOLUTION”) design to account for the design limitations in the SOLUTION study by 1) starting Sollpura dosing as 125% of the pre-study PERT dose, 2) allowing for a more “real life” dose adjustment, as needed, based on signs and symptoms throughout the primary treatment phase of the study, and 3) a shorter treatment duration of 4 weeks, with 3 weeks of dose optimization and 1 week of stable dosing. Patients are then followed in a 20-week extension period for the collection of longer term safety and efficacy (e.g., growth, maintenance of body weight) data. The RESULT study design was discussed with the United States Food and Drug Administration (“FDA”) prior to initiation. Furthermore, the study had been approved by the Cystic Fibrosis Foundation Therapeutics Development Network (“CFFTDN”) Protocol Review Committee, and the European Cystic Fibrosis Society Clinical Trial Network Executive Committee.
The RESULT study enrolled 140 patients in North America, Eastern and Western Europe and Israel. In December 2017 and January 2018, pre-specified interim futility analyses of the RESULT study were conducted by a Data Monitoring Committee (“DMC”) comprised of experts appointed by the CFFTDN when approximately 25% and 50% of patients had completed the 4-week treatment period; in each instance, the committee recommended the study to continue to completion as planned. The company expect to report topline data from the RESULT study in March 2018.
A second, smaller Phase 3 study (“SIMPLICITY”) aimed at expanding the treatment age of patients to include patients age 28 days to seven years old, and enabling potential marketing approval for the sachet presentation of Sollpura, was initiated in the second quarter of 2016. The SIMPLICITY study utilizes sachets containing Sollpura powder for oral solution. The study is designed in two parts (Part A and Part B). Part A which evaluated the safety and general usability of Sollpura powder for oral solution in 15 patients ≥7 years of age, was completed in the fourth quarter of 2016. On December 9, 2016, an independent Data Monitoring Committee evaluated the data from Part A and approved progression to Part B, which will enroll pediatric subjects below 7 years of age. Before the company proceed with Part B, the company plan to amend the SIMPLICITY study to follow a similar dosing approach as in the RESULT study and initiate enrollment in Part B in the second quarter of 2018.
Furthermore, during the third quarter of 2016, the company initiated the EASY study, which provides continued access to Sollpura for patients in the Sollpura arm who completed the SOLUTION study. Anthera Pharmaceuticals has amended this study to also allow Sollpura-assigned patients completing the RESULT study at a lipase dose greater than 10,000 units/kg/day to have continued access until the Biological License Application (“BLA”) for Sollpura is approved by the FDA.
Lastly, prior to the RESULT study, the company conducted another Phase 3 study (“SOLUTION”) in patients with EPI. The SOLUTION study was also a randomized, open-label, assessor-blind, non-inferiority, active-comparator study evaluating the efficacy and safety of Sollpura. This pivotal study enrolled 128 patients in North America, Europe and Israel. Top line data announced in December 2016 showed that the study narrowly missed the CFA non-inferiority margin of the primary mITT analysis by one percent; however, by additional pre-specified analyses of CFA (mITT-Baseline Observation Carried Forward and Per Protocol), Sollpura met the non-inferiority criterion. The study also demonstrated that the ratio of the three enzymes in Sollpura provided an appropriate response in coefficient of nitrogen absorption (“CNA"). In March 2017, the company announced data from the extension phase of the study, which showed that Sollpura demonstrated comparable maintenance in key measurements of height, weight, and body mass index in addition to being well tolerated throughout the 12-week extension period.
The company believe its Sollpura studies may offer a number of potential opportunities for differentiation versus the currently marketed porcine-derived PERTs, including:
- use of biotechnology-derived high-purity enzymes that are produced by fermentation processes rather than from mammalian organs which carry a label warning for viral transmission;
- ability to manufacture at a fixed ratio of lipase, protease and amylase that is similar to the enzyme secretions from the human pancreas;
- use of a novel, chemically-modified lipase drug substance that provides resistance to degradation at gastric pH, thereby obviating the need for enteric coating;
- lack of enteric coating allows for potentially fewer and smaller, easy to swallow capsules and adequate storage stability compared with porcine PERTs of an equivalent unit dose strength; and
- a sachet formulation containing Sollpura powder for oral solution which can be easily dissolved into water, and finally provides patients, especially young pediatric patients, with an easy-to-swallow dosing option.
Blisibimod
BAFF, or B-cell Activating Factor (also known as B lymphocyte stimulator or BLyS), is a member of a tumor necrosis family of natural human proteins and is critical to the development, maintenance and survival of multiple B-cell lineages as well as plasma cells – all of which are critical to the human immune response. B-cells and plasma cells are a vital part of the human immune system, producing natural antibody responses to invading pathogens such as viruses, bacteria and other dangerous antigens. Abnormally high elevations of BAFF, B-cells and plasma cells have been associated with several autoimmune diseases, including lupus and IgA nephropathy. BAFF is primarily expressed by macrophages, monocytes and dendritic cells and interacts with three different receptors on B-cells and plasma cells including BAFF receptor, or BAFF-R, B-cell maturation antigen, or BCMA, and transmembrane activator and cyclophilin ligand interactor, or TACI. The potential role of BAFF inhibition and associated reductions in B-cell and plasma cell numbers in lupus and rheumatoid arthritis has been validated in multiple clinical studies with blisibimod and other BAFF antagonists.
Blisibimod, a peptibody directed against BAFF, was developed as an alternative to antibodies and is produced in Escherichia coli bacterial culture, as opposed to antibodies that are typically produced in mammalian cell culture. A peptibody is a novel fusion protein that is distinct from an antibody with several potential advantages, including ease of manufacture, potency and relatively small molecular weight. Blisibimod inhibits both soluble and membrane-bound BAFF.
The company's Phase 2 Development of Blisibimod for Immunoglobulin A Nephropathy
IgA is a human antibody that helps the body fight infections. IgA nephropathy may occur when plasma cells express excessive amounts of under-glycosylated IgA and subsequent immune complexes containing this immunogenic protein are deposited in the kidneys. These IgA containing immune complexes deposit in the mesangium of glomeruli in the kidney and are proinflammatory. As a result, kidney glomeruli become inflamed and damaged, leading to leakage of blood and protein into urine. According to a recent publication in the New England Journal of Medicine (Wyatt & Julian, 2013), primary IgA nephropathy occurs at any age, most commonly with clinical onset in the second and third decades of life, and a large number of cases eventually progress to renal failure. In patients with IgA nephropathy, levels of BAFF are significantly higher than in healthy individuals, and elevated levels of BAFF are associated with histological severity of disease in kidney tissue. In IgA nephropathy, plasma cells express immunogenic IgA that forms immune complexes that deposit in renal tissue and lead to renal inflammation and damage that can progress to renal failure and end-stage renal disease. Significant reductions in B-cell counts were observed in clinical studies of patients with lupus with blisibimod with concomitant decreases in proteinuria, serum immunoglobulins and autoantibodies, and increases in complement C3. The company believe inhibition of BAFF may reduce B-cell proliferation, maturation, and survival, thereby reducing serum levels of IgA and targeting antibodies, and therefore reduce progressive renal damage in patients with IgA nephropathy.
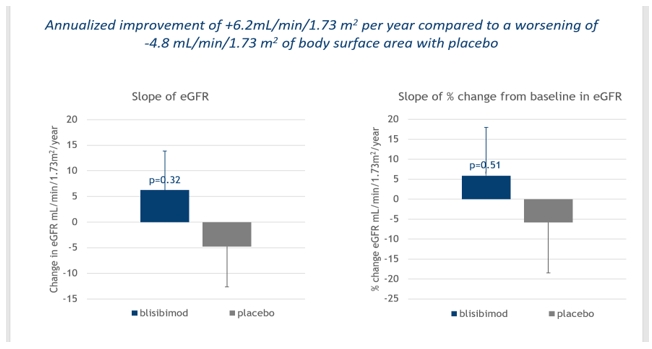
In June 2013, the company initiated a Phase 2 clinical study, (“BRIGHT-SC”) of patients with IgA nephropathy in Asia and Eastern Europe. The BRIGHT-SC study was a Phase 2 multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy, safety, tolerability and immunogenicity of blisibimod in IgA nephropathy. Enrollment criteria included biopsy-proven IgA nephropathy and proteinuria greater than one gram but less than six grams per 24 hours (1g-6g/24hr). Patients must have been receiving standard of care medication including angiotensin converting enzyme inhibitors and/or angiotensin receptor blockers. Patients enrolled in the BRIGHT-SC study received 300mg weekly blisibimod or placebo subcutaneously during the first 8 weeks of therapy, the induction phase, followed by a minimum of 24 weeks of 200mg weekly blisibimod or placebo, the maintenance phase. The BRIGHT-SC study enrolled 58 patients. In August 2017, the company reported top line data from the completed extension of the BRIGHT-SC study in which all patients had the opportunity to complete at least 60 weeks of treatment and some patients were treated for up to two years. Throughout the treatment period and for up to one year of additional follow up off treatment, blisibimod appeared to halt disease progression as measured by the mean estimate of urinary protein:creatinine levels ("proteinuria"). Specifically, in patients treated with blisibimod, the mean change in proteinuria was stable to trending slightly downward, whereas the mean levels increased for patients in the placebo arm. Additionally, blisibimod showed a trend toward preservation of renal function based upon individual rates of change in estimated glomerular filtration rate (“eGFR”), with an annualized improvement of +6.2mL/min/1.73 m2 per year compared to a worsening of -4.8 mL/min/1.73 m2 with placebo as seen in the graph below. Furthermore, serum immunoglobulins IgA, IgG, and IgM, demonstrated marked reduction throughout the treatment period.
Market Opportunity
Sollpura for the treatment of Exocrine Pancreatic Insufficiency (EPI)
According to its estimate, EPI is a disease that affects an estimated 130,000 patients in the United States. The most common causes of EPI are chronic pancreatitis and cystic fibrosis, the former a longstanding inflammation of the pancreas altering the organ's normal structure and function that can result from malnutrition, heredity, or (in the western world especially), behavior (alcohol use and smoking), and the latter a recessive hereditary disease most common in Europeans and Ashkenazi Jews where the molecular culprit is an altered, CFTR-encoded chloride channel. In children, another common cause is Shwachman-Bodian-Diamond syndrome, a rare autosomal recessive genetic disorder resulting from mutation in the SBDS gene.
Blisibimod for the treatment of IgA Nephropathy
According to the National Organization for Rare Diseases, primary IgA nephropathy occurs at any age, most commonly with clinical onset in the second and third decades of life and a large number of cases eventually progress to renal failure. There is also a striking geographic variation in the prevalence of IgA nephropathy throughout the world. In the United States, IgA nephropathy is considered an orphan disease as it is believed to affect approximately 130,000 people annually. In August 2017, the FDA granted orphan drug designation for blisibimod for the treatment of IgA. The prevalence of IgA nephropathy varies throughout the world, with the highest prevalence in Asia (Singapore, Japan and China), Australia, Finland and southern Europe (20 to 40% of all glomerulonephritis). In Asia, routine urinalyses are performed for school children and renal biopsies for patients with asymptomatic hematuria, and the reported prevalence of the disease is much higher. For example, in Japan, IgA nephropathy is estimated to affect over 350,000 people annually. According to the National Kidney and Urologic Diseases Information Clearinghouse, 25% of adults with IgA nephropathy eventually develop total kidney failure.
Manufacturing Strategy
Sollpura
The company completed technology transfer for pharmaceutical ingredient (“API”) manufacturing and drug product manufacturing in 2016, and estimate that the company will complete process validation for the fermentation and associated down-stream purification for all three enzyme APIs in 2019. In parallel, commercial scale capsule and sachet drug product manufacturing process validation is anticipated to be completed in 2019.
Regulatory Strategy
Sollpura
The RESULT study protocol has been reviewed by the FDA prior to initiation. The company believe that the design of RESULT provides adequate evaluation of efficacy and safety of Sollpura to respond to the FDA’s 2011 complete response letter. The RESULT study should also address the 2005 EMA protocol assistance comments, which were consistent with the FDA’s request for an active comparator trial. The company anticipate completing the Phase 3 clinical trials with Sollpura (the RESULT and SIMPLICITY studies) in 2018, and process validation for commercial manufacture of Sollpura APIs and drug product in 2019, with a target submission date of the Biologics Licence Application (“BLA”) in 2019.
IgA Nephropathy
In September 2013, the company met with the FDA who agreed to consider accepting proteinuria as a surrogate endpoint under a Subpart E approval for blisibimod for treatment of IgA nephropathy. In April 2014, the company met with the Japan Pharmaceuticals and Medical Devices Agency (“PMDA”) to discuss its registration program for blisibimod in IgA nephropathy. In this meeting the company gained the PMDA’s agreement on the acceptability of proteinuria as the primary efficacy endpoint to support marketing approval in Japan. In December 2014 the company met with the European Medicines Agency (“EMA”) as part of the scientific advice process for blisibimod, and reached agreement on the acceptability of proteinuria as the primary efficacy variable, as well as the sufficiency of a single study to support a Conditional Marketing Authorization Application (“CMAA”) provided that confirmatory evidence from a second study would be available post approval. The EMA also recommended that the protocol provide information on the required duration of treatment, duration of response and need for re-treatment. Given the observed effects of blisibimod on proteinuria in patients with lupus, Anthera Pharmaceuticals is currently evaluating its options for investigating the effects of blisibimod in B-cell associated glomerulonephritides.
Historical Clinical Studies by Licensor - Sollpura
Sollpura was studied from 2002 to 2009 in seven clinical trials, in which a total of 492 unique subjects received at least 1 dose of Sollpura. Three Phase 1 trials were conducted, 1 in healthy volunteers and 2 in subjects with EPI due to CF. Two short-term trials, the Phase 2 Study TC-2A, and the Phase 3 Study 726 evaluated the efficacy of Sollpura in subjects ≥7 years of age with EPI due to CF. Two long-term Phase 3 safety and tolerability trials were also conducted: Study 767 in subjects with EPI due to CF and Study 810 in subjects with EPI due to chronic pancreatitis/pancreatectomy. Completed clinical trials demonstrated that dietary fat and nitrogen (protein) absorption are significantly increased in patients with cystic fibrosis and EPI who received Sollpura. In 2013, Eli Lilly gained agreement from the FDA on the design of a pivotal trial that would provide adequate evaluation of efficacy and safety.
The dose-ranging Phase 1 study TC-1B evaluated five dose levels across a 50-fold range, from 100-to-5000 lipase units (U) per kg per meal in CF-EPI subjects. In this study, greater improvements in nutrients absorption, as measured using the percent change from baseline in the coefficient of fat absorption (CFA) and CNA, were observed at doses of 500 lipase U per kg per meal and higher.
Study TC-2A, a Phase 2, randomized, double-blind, parallel group, dose-finding trial, was conducted in 125 pediatric and adult subjects with CF-related EPI who were treated with Sollpura for 28 days in one of three dosing regimens containing 6,500 U, 32,500 U, and 130,000 U lipase administered per meal or snack. Observed mean CFAs at the end of study were 56.2%, 67.0%, and 69.7% in the 6,500, 32,500, and 130,000 U dose groups, respectively (one-way ANOVA p = 0.0032) with significant improvements in mean changes from baseline (one-way ANOVA p = 0.0005) and mean changes from baseline off-enzyme CFA to on treatment were 1.2%, 11.4% and 17.3%, respectively (1-way ANOVA p=0.0005). Pairwise comparison of the CFA values showed that statistically greater improvements were observed at the higher doses of Sollpura compared with the lowest dose of 6,500 U. Similar improvements in the CNA, were observed with Sollpura at the two highest dose levels.
Study 726, a Phase 3, placebo-controlled, parallel design, multinational clinical, evaluated the effects of a single capsule of Sollpura (containing 32,500 U lipase, with protease and amylase in fixed ratios) or placebo administered with every meal or snack in subjects with cystic fibrosis-related EPI. Among the 138 subjects enrolled in this study, treatment with Sollpura resulted in a statistically significant improvement in the change from baseline in the CFA of 21.2% with Sollpura compared with 6.0% with placebo (p = 0.0011). The median body weight of subjects in this trial was 50 kg, for which the corresponding Sollpura dose was 650 U lipase/kg/meal or snack.
Two one-year studies were conducted to evaluate the safety and effects on nutritional status of Sollpura. Study 810 evaluated adult subjects with EPI due to CP or after pancreatectomy. Among the 214 subjects who were treated, an average dose of 5.5 capsules (containing 32,500 U lipase, with protease and amylase in fixed ratios) of Sollpura per day maintained nutritional status as assessed by serial measurement of height and weight, including age-appropriate growth and weight gain in children. Mean BMI z-scores for subjects in Study 767 were maintained over time on study (mean BMI z-score at baseline, Months 3, 6, and 12 were -0.503, -0.637, -0.688, and -0.655, respectively).
Historical Clinical Studies – Blisibimod
To date, five randomized, clinical studies have been conducted with blisibimod in patients with lupus: two Phase 1 dose-ranging studies by its licensor, Amgen; and two double-blind, placebo-controlled dose-ranging clinical outcomes studies, PEARL-SC (Phase 2b) and CHABLIS-SC1 (Phase 3), and a Phase 2 Open-Label Extension study (OLE) by the Company. All five clinical studies evaluated the efficacy and safety of multiple doses of subcutaneous blisibimod versus placebo in patients with active and seropositive lupus. The PEARL-SC study was completed in June 2012 and the CHABLIS-SC1 study was completed in November 2016. Both studies failed to meet the primary endpoints and the company elected to stop further development of blisibimod for the treatment of lupus.
In the CHABLIS-SC1 study, the primary endpoint compared the effects of blisibimod and placebo at Week 52 using the SLE Responder Index-6 (SRI-6): ≥6-point improvement in SELENA-SLEDAI, no new BILAG 1A or 2B domain scores, and <0.3-point increase in Physician’s Global Assessment. Compared with placebo, a higher proportion of subjects on blisibimod met the SRI-6 criteria from Week 16 through Week 52. However, this effect was not statistically significant at the primary endpoint analysis at Week 52. There was a statistically-significant steroid sparing effect among subjects randomized to blisibimod wherein 17.2% of subjects in the blisibimod group achieved corticosteroid dose of less than or equivalent to 7.5 mg prednisone compared with 8.9% in the control group (p=0.019). In subjects with baseline urinary protein:creatinine ratio (UPCR) ≥0.5 g/g, significantly higher proportions of blisibimod subjects achieved >50% reduction in UPCR, and/or UPCR <0.5 g/g. Reductions in SLE autoantibodies and B cells, and increases in complement C3 and C4 were observed with blisibimod. The company believe that these data support the further research with blisibimod patients with B-cell associated glomerulonephritides including IgA nephropathy, lupus with renal manifestations, and membranous glomerulonephritis. As a result of the outcomes of the CHABLIS-SC1 trial, at the end of 2016 the company elected to discontinue the Phase 3 CHABLIS-7.5 study, which was initiated in June 2016.
Research and Development
Since its inception in 2004, Anthera Pharmaceuticals has focused primarily on developing its product candidates, which currently include Sollpura for EPI and blisibimod for IgA nephropathy and potentially other glomerulonephritides. In the years ended December 31, 2017, 2016, and 2015, the company incurred $28.6 million, $46.5 million and $33.5 million, respectively, of research and development expense.
Strategy
The company's objective is to develop and commercialize its product candidates to treat serious diseases associated with inflammation, including enzyme replacement therapies and renal disease. To achieve these objectives, the company intend to initially focus on the following activities.
Advance Clinical Development of Sollpura
Anthera Pharmaceuticals is advancing the development of Sollpura in a Phase 3 registration program in patients with cystic fibrosis-related EPI. If its Phase 3 clinical study is successful, the company intend to commercialize Sollpura in the U.S. and seek strategic corporate partners whose capabilities complement ours to launch Sollpura outside of U.S.
Seek Collaborative Corporate Partner for Blisibimod
Anthera Pharmaceuticals has received orphan drug designation for blisibimod for the treatment of IgA nephropathy. The company plan to opportunistically enter into collaborations with third parties for the development of blisibimod in renal disease and other B-cell associated glomerulonephritides.
Developing Commercial Strategies Designed to Maximize The company's Product Candidates’ Market Potential
The company's product candidates are focused on highly-specialized physician segments, such as cystic fibrosis specialists and nephrologists. The company believe that the company can build a small, focused sales force capable of marketing its products effectively in acute care and orphan indications.
Competition
The company's industry is highly competitive and subject to rapid and significant technological change. The company's potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and research institutions. The company's primary competitors who market PERTs approved in the U.S. or are developing PERTs in the U.S. are described in further detail below. The company believe that key competitive factors that will affect the development and commercial success of its product candidates are efficacy, safety and tolerability profile, reliability, convenience of dosing, price and reimbursement.
| Compound | Stage | Company | Indications | Notes |
|---|---|---|---|---|
| Creon | Approved | Abbvie | EPI, CF, CP, pancreatectomy | Ÿ Porcine, enteric coated |
| Pancreaze | Approved | Janssen/J&J | EPI, CF and other | Ÿ Porcine, enteric coated |
| Zenpep | Approved | Allergan | EPI, CF, CP | Ÿ Porcine, enteric coated |
| Viokace | Approved | Allergan | EPI, CP and pancreatectomy in adults | Ÿ Porcine, enteric coated, in combination with proton pump inhibitor |
| Pertzye | Approved | Chiesi | EPI, CF | Ÿ Porcine, enteric coated |
| MS1819 | Phase 2a | AzurRx | EPI, CP | Ÿ Lipase only |
Intellectual Property
The company's policy is to pursue, maintain and defend patent rights, developed internally and licensed from third parties, to protect the technology, inventions and improvements that are commercially important to the development of its business. The company also rely on trade secrets that may be important to the development of its business.
The company's success will depend significantly on its ability to:
- obtain and maintain patent and other proprietary protection for the technology, inventions and improvements the company consider important to its business;
- defend its patents;
- preserve the confidentiality of its trade secrets; and
- operate its business without infringing the patents and proprietary rights of third parties.
Sollpura
As of the date of this report, its Sollpura portfolio is made up of exclusively licensed patents and patent applications from Eli Lilly, including:
- Two issued U.S. patents;
- Four issued European (“EP”) patents, each validated in one or more of Austria, Belgium, Bulgaria, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Great Britain, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Monaco, Netherlands, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, Switzerland and Turkey;
- 16 issued non-EP foreign patents in Australia, Canada, China, Hong Kong, India, Israel, Japan, Mexico, Russia, South Korea and Ukraine; and
- One pending non-EP foreign patent application.
The company hold exclusive worldwide licenses from Eli Lilly to all of these patents and patent applications. The exclusively licensed U.S. patents are currently scheduled to expire in March 2025 and July 2028. Depending upon the timing, duration and specifics of FDA approval of Sollpura, one of these U.S. patents may be eligible for a patent term restoration of up to five years under Hatch-Waxman Act. See “—Regulatory Matters— Patent Term Restoration and Marketing Exclusivity.” This could extend the expiration date of the selected U.S. Patent to as late as March 2030 or July 2033, depending on which patent the term restoration is applied to. The company intend to pursue pediatric exclusivity as well, which could add an additional six months to the patent term. The four exclusively licensed EP patents are currently scheduled to expire between February 2021 and October 2025. One of these patents may be eligible for a Supplementary Protection Certificate of up to five years, which could extend the expiration date to between February 2026 and October 2030.
Blisibimod
As of the date of this report, its blisibimod patent portfolio includes:
- Four issued U.S. patents;
- One pending U.S. non-provisional patent application;
- Three issued European (EP) patents, each validated in one or more of Albania, Austria, Belgium, Cyprus, Denmark, Finland, France, Germany, Greece, Ireland, Italy, Latvia, Liechtenstein, Lithuania, Luxembourg, Monaco, the Netherlands, Portugal, Romania, Slovenia, Spain, Sweden, Switzerland, Turkey and the United Kingdom;
- One pending EP patent application;
- 23 issued non-EP foreign patents in Australia, Bulgaria, Canada, China, the Czech Republic, Estonia, Eurasia (validated in all nine Eurasian countries), Hong Kong, Hungary, Israel, Japan, Mexico, New Zealand, Norway, the Philippines, Poland, Serbia, Singapore, Slovakia, South Korea and South Africa; and
- Four pending non-EP foreign patent applications in Brazil, Hong Kong, Mexico, and Poland.
The company hold exclusive worldwide licenses from Amgen to all of these patents and patent applications. In addition, the company hold a non-exclusive worldwide license to one issued U.S. patent, one pending U.S. non-provisional patent application, one EP patent, one pending EP patent application, and over 50 non-EP foreign patents and pending patent applications relating to general peptibody compositions and formulations.
The four exclusively licensed U.S. patents are currently scheduled to expire in May 2022, March 2023 and November 2023. Depending upon the timing, duration and specifics of FDA approval of blisibimod, one of these U.S. patents (or another patent issuing from a related patent application) is expected to be eligible for a patent term restoration of up to five years under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act. See “—Regulatory Matters— Patent Term Restoration and Marketing Exclusivity.” This could extend the expiration date of the U.S. Patent to as late as May 2027, March 2028 or November 2028, depending on which patent the term restoration is applied to. The company intend to pursue pediatric exclusivity as well, which could add an additional six months to the patent term. The exclusively licensed EP patents are currently scheduled to expire in May 2022. One of these patents is expected to be eligible for a Supplementary Protection Certificate of up to five years, which could extend the expiration date to May 2027.
The U.S. patent system permits the filing of provisional and non-provisional patent applications. A non-provisional patent application is examined by the United States Patent and Trademark Office, or USPTO, and can mature into a patent once the USPTO determines that the claimed invention meets the standards for patentability. A provisional patent application is not examined, and automatically expires 12 months after its filing date. As a result, a provisional patent application cannot mature into a patent. The requirements for filing a provisional patent application are not as strict as those for filing a non-provisional patent application. Provisional applications are often used, among other things, to establish an early filing date for a subsequent non-provisional patent application.
The filing date of a non-provisional patent application is used by the USPTO to determine what information is prior art when it considers the patentability of a claimed invention. If certain requirements are satisfied, a non-provisional patent application can claim the benefit of the filing date of an earlier filed provisional patent application. As a result, the filing date accorded by the provisional patent application may remove information that otherwise could preclude the patentability of an invention.
Anthera Pharmaceuticals is aware of two third-party issued U.S. patents that contain broad claims related to BLyS or BAFF binding polypeptides. Based on its analyses, if these patents were asserted against it, the company do not believe that blisibimod would be found to infringe any valid claim of these patents. If the company were to challenge the validity of either of these issued U.S. patents in court, the company would need to overcome the presumption of validity that attaches to every U.S. patent by presenting clear and convincing evidence as to the invalidity of the patent’s claims. There is no assurance that a court would find in its favor on questions of infringement or validity, and the company could incur substantial costs in litigation if Anthera Pharmaceuticals is required to defend against patent suits brought by third parties or if the company initiate these suits. If third-party patents are determined to be valid and construed to cover blisibimod, the development and commercialization of this program could be affected, subjecting it to potential liability for damages and possibly requiring it to obtain a license to continue marketing the affected product. Such a license may not be available on commercially acceptable terms, if at all.
Current License Agreements
Eli Lilly and Company
In July 2014, the company entered into a worldwide, exclusive license agreement with Eli Lilly (the “Lilly Agreement”), to develop and commercialize Sollpura, a Phase 3 novel investigational PERT for the treatment of patients with EPI, often seen in patients with cystic fibrosis and other conditions. Under the terms of the Lilly Agreement, the company were not required to make any up-front payment but are obligated to make milestone payments of up to $33.5 million for capsule products and $9.5 million for reformulated products upon the achievement of certain regulatory and commercial sales milestones, none of which have been achieved as of December 31, 2017. In addition, after sales of the licensed products exceed an aggregate of $100.0 million in the United States, Anthera Pharmaceuticals is obligated to pay tiered royalties on future net sales of products, ranging from the single digits to the mid-teens for products that are developed and approved as defined in the Lilly Agreement. The company's royalty obligations as to a particular licensed product will be payable, on a licensed product-by-licensed product basis, for the longer of (a) the date of expiration of the last to expire valid claim within the licensed patents that covers the manufacture, use or sale, offer to sell, or import of such licensed product by it or a sublicense in such country, or (b) 12 years after the first commercial sale of the applicable licensed product in the applicable country.
Amgen
In December 2007, the company entered into a license agreement with Amgen, which was amended in October 2009 and November 2014 (as amended, the “Amgen Agreement”), pursuant to which the company obtained an exclusive worldwide license to certain technology and compounds relating to blisibimod, as well as a non-exclusive worldwide license to technology relating to certain peptibody compositions of matter and formulations. The licensed patents included a specific set of previously filed U.S. and foreign patents and applications, as well as any applications filed after the execution date by Amgen and covering licensed know-how. During the period of the agreement, Anthera Pharmaceuticals is responsible for the filing, prosecution, defense and maintenance of all exclusively licensed blisibimod patents and applications. Amgen retains the right to review all documents relating to said filing, prosecution, defense and maintenance, and Anthera Pharmaceuticals is required to incorporate all reasonable comments or suggestions that Amgen makes with regard to these documents.
Pursuant to the terms of the Amgen Agreement, Anthera Pharmaceuticals has paid $6.0 million in license fees to Amgen for blisibimod. In addition, Anthera Pharmaceuticals is required to make various milestone payments upon the achievement of certain development, regulatory and commercial objectives, including payment upon commencement of the first Phase 3 clinical study for any blisibimod formulation in the United States or European Union. Anthera Pharmaceuticals is also required to pay up to $10.0 million upon achievement of certain pre-approval clinical development milestones and up to $23.0 million upon achievement of certain post-approval milestones. Furthermore, Anthera Pharmaceuticals is required to make tiered quarterly royalty payments on net sales, which increase as a percentage from the high single digits to the low double digits as net sales increase. The company's royalty payment obligations for a particular product in a particular country begin on the date of the first commercial sale of the licensed product in that country, and end upon the later of 10 years from the date of first commercial sale in that country or the expiration date of the last valid claim of a licensed patent that covers the manufacture, use or sale, offer to sell or import of the product.
The Amgen Agreement will remain in effect until the company elect to terminate, or until termination for material breach by either party or insolvency on its part. Under these terms, Amgen can terminate the agreement if the company fail to meet its obligations, resulting in a loss of its exclusive rights to the licensed technology.
In connection with a collaborative arrangement with Zenyaku Kogyo Co., Ltd (“Zenyaku”) for the development of IgA nephropathy that was executed in December 2014 and terminated in January 2016, the company amended the Amgen Agreement in November 2014 to ![]() adjust certain royalty and milestone payment obligations payable to Amgen in light of its collaboration with Zenyaku and (ii) provide that the sublicense granted by it to Zenyaku shall survive the termination of the Amgen Agreement. Under this amendment, the company also agreed to grant Amgen that number of shares of its common stock equal to $1.0 million divided by the volume weighted average price of its common stock for 20 trading days prior to issuance. The company issued 420,751 shares of common stock to Amgen at $2.3767 per share on January 28, 2015 pursuant to a subscription agreement with Amgen, with the consideration paid by Amgen in the form of a waiver of a fee otherwise payable to Amgen under the Amgen Agreement.
adjust certain royalty and milestone payment obligations payable to Amgen in light of its collaboration with Zenyaku and (ii) provide that the sublicense granted by it to Zenyaku shall survive the termination of the Amgen Agreement. Under this amendment, the company also agreed to grant Amgen that number of shares of its common stock equal to $1.0 million divided by the volume weighted average price of its common stock for 20 trading days prior to issuance. The company issued 420,751 shares of common stock to Amgen at $2.3767 per share on January 28, 2015 pursuant to a subscription agreement with Amgen, with the consideration paid by Amgen in the form of a waiver of a fee otherwise payable to Amgen under the Amgen Agreement.
Manufacturing and Supply
The company currently rely on contract manufacturers to produce drug substances and drug products required for its clinical studies under current GMP with oversight by its internal managers. The company plan to continue to rely upon contract manufacturers and, potentially, collaboration partners to manufacture commercial quantities of its product candidates if and when approved for marketing by the FDA. The company's contract manufacturers obtain the raw materials for the drug substances and drug products required for its clinical studies from a variety of sources. The company believe that this will provide a sufficient supply of these raw materials and drug product to meet its needs for the foreseeable future. The company do not have in place long term supply agreements with respect to all of the components of any of its pharmaceutical systems, however, and are subject to the risk that the company may not be able to procure all required components in adequate quantities with acceptable quality, within acceptable time frames or at reasonable cost.
The company's research and development activities involve the controlled use of potentially hazardous substances, including toxic chemical and biological materials. Accordingly, Anthera Pharmaceuticals is subject to federal, state and local laws governing the use, handling and disposal of these materials. The company believe that its safety procedures for handling and disposing of these materials comply in all material respects with the standards prescribed by local, state and federal regulations.
Sales and Marketing
Given its stage of development, Anthera Pharmaceuticals has not developed a commercial organization or distribution capabilities. The company expect that the company would develop these capabilities once the company receive Phase 3 data in contemplation of FDA approval and the commercial launch of its product candidates. In order to commercialize its product candidates, the company plan to develop these capabilities internally or through collaboration with third parties. In selected therapeutic areas where the company feel that any approved products can be commercialized by a specialty sales force that calls on a limited and focused group of physicians, the company may seek to commercialize the product candidates alone. The company also plan to seek commercialization partners for products in international markets.
The company intend to build the commercial infrastructure necessary to bring its product candidates to market. In addition to a specialty sales force, sales management, internal sales support and an internal marketing group, the company will need to establish capabilities to manage key accounts, such as managed care organizations, group-purchasing organizations, specialty pharmacies and government accounts. The company may also choose to employ medical sales liaisons personnel to support its products.




