Catabasis Pharmaceuticals
Overview
Catabasis Pharmaceuticals (CATB) is a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of novel therapeutics based on its proprietary Safely Metabolized And Rationally Targeted linker, or SMART LinkerSM, drug discovery platform. The company's SMART Linker drug discovery platform enables it to engineer product candidates that can simultaneously modulate multiple targets in a disease. The company's proprietary product candidates impact pathways that are central to diseases where efficacy may be optimized by a multiple target approach. Catabasis Pharmaceuticals has applied its SMART Linker drug discovery platform to build an internal pipeline of product candidates for rare diseases, its primary focus, and plan to pursue partnerships to develop additional product candidates.1
The company's lead product candidate is edasalonexent, formerly known as CAT-1004, which the company believe has the potential to be a disease-modifying therapy for all patients affected by Duchenne muscular dystrophy, or DMD, regardless of the underlying dystrophin mutation. Edasalonexent is an oral small molecule that inhibits NF-kB, or nuclear factor kappa-light-chain-enhancer of activated B cells. DMD is an ultimately fatal genetic disorder involving progressive muscle degeneration. The United States Food and Drug Administration, or FDA, has granted orphan drug, fast track and rare pediatric disease designations to edasalonexent for the treatment of DMD. The European Commission, or EC, has granted orphan medicinal product designation to edasalonexent for the treatment of DMD.
The company's MoveDMD® Phase 1/2 trial enrolled ambulatory boys four to seven years old with a genetically confirmed diagnosis of DMD who were steroid naive or had not used steroids for at least six months prior to the trial. Boys enrolled in the trial were not limited to any specific dystrophin mutations, and the 31 boys in the trial had 26 different dystrophin mutations. The MoveDMD trial was designed to be conducted in three sequential parts, Phase 1, Phase 2, and an open-label extension, which is on-going.
Catabasis Pharmaceuticals has completed key efficacy and safety assessments from the MoveDMD trial. In the open-label extension of the MoveDMD trial after more than a year of oral 100 mg/kg/day edasalonexent treatment, the company observed preserved muscle function and consistent improvements in all four assessments of muscle function compared to the rates of change in the control period for boys prior to receiving edasalonexent treatment. Additionally, supportive changes in non-effort based measures of muscle health were seen, supporting the durability of edasalonexent treatment effects. Through more than one year of treatment, edasalonexent continued to be well tolerated with no safety signals observed in the trial. The company plan to initiate a global Phase 3 trial in DMD in the first half of 2018 to evaluate the efficacy and safety of edasalonexent for registration purposes and expect to report top-line results from this trial in 2020, dependent on raising capital.
In addition to edasalonexent, Catabasis Pharmaceuticals is developing a pipeline of product candidates using its SMART Linker drug discovery platform as potential treatments for rare diseases. The company's pipeline includes CAT-5571, which Catabasis Pharmaceuticals is developing as a potential treatment for cystic fibrosis, or CF, and CAT-4001, which Catabasis Pharmaceuticals is developing as a potential treatment for neurodegenerative diseases such as amyotrophic lateral sclerosis, or ALS, and Friedreich's ataxia, or FA. CAT-5571 is a small molecule that is designed to restore host defense by activating autophagy, a mechanism for recycling cellular components and digesting pathogens, which is depressed in CF. CAT-4001 is a small molecule that activates Nuclear factor (erythroid-derived 2)-like 2, or Nrf2, and inhibits NF-kB, two pathways that have been implicated in FA and ALS. The company plan to complete investigational new drug, or IND, application-enabling activities for CAT-5571 and to advance CAT-5571 into a Phase 1 clinical trial in the second half 2018 and report top-line results in 2019, based on its current operating plan.
Catabasis Pharmaceuticals has previously applied its SMART Linker drug discovery platform to engineer its CAT-2000 series product candidates to inhibit the Sterol Regulatory Element Binding Protein, or SREBP, pathway. Catabasis Pharmaceuticals has advanced two CAT-2000 molecules, CAT-2003 and CAT-2054, into clinical development and intend to pursue a partnership for further development of the CAT-2000 series for the treatment of nonalcoholic steatohepatitis, or NASH.
As of December 31, 2017, the company had six issued U.S. patents with composition of matter and method of use claims directed to edasalonexent, six issued U.S. patents with composition of matter and method of use claims directed to the CAT-2000 series, four issued U.S. patents with composition of matter and method of use claims directed to CAT-5571 and two issued U.S. patents with composition of matter and method of use claims directed to CAT-4001. These patents are expected to expire between 2029 and 2031, without taking into account potential patent term extensions. In addition, its patent portfolio includes over 100 issued foreign patents, over 10 pending U.S. patent applications and over 35 pending foreign patent applications.
The company's Scientific Approach and SMART Linker Drug Discovery Platform
The company's SMART Linker drug discovery platform enables it to engineer product candidates that can simultaneously modulate multiple biological targets in a disease. The company's proprietary product candidates impact pathways that are central to diseases where efficacy may be optimized by a multiple target approach. The company's aim is to leverage the growing body of knowledge associated with disease pathways, and to rationally design orally bioavailable product candidates that simultaneously interact with multiple biological targets in a disease. While other technologies exist to conjugate or combine two bioactives, the company believe that its SMART Linker drug discovery platform provides substantial improvements over previous approaches to bioactive conjugation.
The company's SMART Linker drug discovery platform includes a broad array of linkers that the company use to engineer molecular series that can simultaneously modulate multiple biological targets in a disease. The linkers used in its drug discovery platform are small chemicals designed to join two separate bioactives into a single conjugate molecule, and some linkers are also bioactives. In systemic circulation, its SMART Linker conjugates are typically stable and inactive, potentially reducing off-target toxicities and side-effects. Certain of its conjugates are designed to be cleaved by specific enzymes exclusively within cells in order to release the two bioactives inside the cells. By releasing the bioactive components of the conjugate molecule inside cells, the SMART Linker allows the bioactives to reach their targets more efficiently and have greater efficacy than if the bioactives were dosed independently or in combination.
Catabasis Pharmaceuticals has SMART Linker conjugates that are designed to be stable with oral dosing, as well as stable in both the lumen of the intestine and in systemic circulation, which Catabasis Pharmaceuticals has now observed in clinical trials for two product candidate series. The company can design the SMART Linker to chemically link the two bioactive molecules through their pharmacophores, the regions of the bioactive molecules that are responsible for carrying out their biological activity, resulting in inactivation of the bioactives while conjugated. Once the conjugate enters a cell, the SMART Linker may be cleaved by specific enzymes which reside only within cells, releasing the two bioactives to interact with their biological targets. Simultaneous delivery of the bioactives through the SMART Linker conjugate into the cell results in the two bioactives having the same pharmacokinetics and tissue distribution. As a result, its SMART Linker conjugates can simultaneously modulate two biological targets in diseases of interest within the same cell. In addition, release of the bioactives inside cells can potentially reduce or eliminate off-target, extracellular activity of the bioactives, which may improve safety and tolerability.
Catabasis Pharmaceuticals has observed in multiple preclinical studies that its SMART Linker conjugates achieved greater efficacy than administration of the two bioactives either independently or in combination. In clinical trials, SMART Linker conjugates have demonstrated significant improvements in activity on disease pathways and tolerability relative to equivalent doses of the two bioactives delivered in combination. The company also have observed statistically significant pharmacological effects with SMART Linker conjugates at dose levels significantly lower than the prescribed doses of the two component bioactives, as further described below under "—The company's Product Candidates—Edasalonexent—Edasalonexent Clinical Development—Completed Clinical Trials".
Product Candidates
The following chart summarizes key information regarding its product candidates. The company hold rights to all of its product candidates throughout the world.
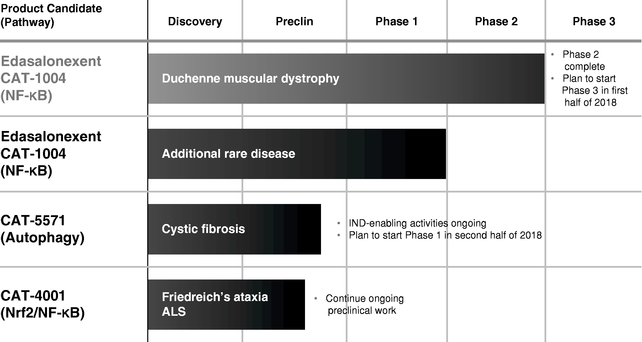
Edasalonexent
Edasalonexent is a SMART Linker conjugate of salicylic acid and the omega-3 fatty acid docosahexaenoic acid, or DHA, a naturally occurring unsaturated fatty acid with anti-inflammatory properties. The company designed edasalonexent to inhibit NF-kB, a protein that is activated in DMD and that drives inflammation, fibrosis and muscle degeneration, and suppresses muscle regeneration. Catabasis Pharmaceuticals has reported results from Phase 1, Phase 2 and the open-label extension of the MoveDMD trial through administration of edasalonexent for up to 60 weeks, as described further below under "—Edasalonexent Clinical Development". The FDA has granted edasalonexent orphan drug, fast track and rare pediatric disease designations for the treatment of DMD. The EC has granted orphan medicinal product designation to edasalonexent for the treatment of DMD.
Overview of DMD
DMD is a rare pediatric disorder involving progressive muscle degeneration that eventually leads to death. DMD is caused by various mutations in the dystrophin gene that result in a lack of functional dystrophin in muscle fibers, which renders muscle fibers more susceptible to mechanical stress. Dystrophin is a protein that resides in the membrane of muscle cells and is critical to the structural and membrane stability of muscle fibers in skeletal, diaphragm, and cardiac muscle. When muscles contract or stretch during normal use, the absence of normally functioning dystrophin results in activation of the NF-kB pathway, triggering inflammation in the muscles, initiating muscle degeneration, and reducing the ability of muscles to regenerate. As muscle damage progresses, connective and adipose tissues replace muscle fibers, resulting in inexorable muscle weakness.
DMD occurs almost exclusively in males, occurring in approximately 1 in 3,500 live male births. Based on this incidence rate, the company estimate that DMD affects a total of approximately 15,000 patients in the United States and approximately 19,000 patients in the European Union.
Children with DMD typically begin to show symptoms of disease between ages two and five, when they develop a waddling gait, frequently fall and have difficulty rising from the floor. Progressive weakness then develops in the voluntary muscles in the arms, legs and trunk. This muscle weakness is accompanied by fixations, or contractures, of joints, such as knees, hips and elbows. By age eight, most patients have difficulty ascending stairs. Patients typically lose walking ability between the ages of ten and fourteen and, by about twelve years of age, most people with DMD are unable to walk and need to use a power wheelchair on a regular basis. Patients' cardiac and respiratory muscles are also adversely affected, typically requiring use of ventilators in their late teens. Progressive weakening of cardiac and respiratory muscles of DMD patients eventually results in death, generally in the patient's mid-twenties.
The Role of NF-kB in Duchenne Muscular Dystrophy
NF-kB plays an important role in regulating skeletal muscle health and appears to be especially important in regulating skeletal muscle mass in chronic diseases such as DMD. Activated NF-kB promotes the degradation of specific muscle proteins and leads to the induction of pro-inflammatory mediators such as cytokines, including tumor necrosis factor alpha, or TNF-a, interleukin 6, or IL-6, and interleukin-1 beta, or IL-1b; chemokines; cell adhesion molecules; and tissue degrading enzymes, such as matrix metallopeptidase 9, or MMP-9. In addition, activated NF-kB suppresses muscle stem cell differentiation that is required for muscle regeneration by preventing satellite stem cells from differentiating into myoblasts, progenitor cells that differentiate, to give rise to muscle cells. Activation of NF-kB is observed in muscle tissues of patients with DMD prior to the onset of other clinical manifestations, and activated NF-kB is persistently elevated in the immune cells and regenerative muscle fibers of patients with DMD. Moreover, evidence exists that mechanical stress activates NF-kB in muscles and increases levels of activated NF-kB by a factor of three to four times. Muscles with increased mechanical stress, such as quadriceps and hamstrings, show the most rapid progression of disease.
Unaddressed Market Opportunity
There are currently only two therapies approved in the United States for the treatment of DMD: the Sarepta Therapeutics, Inc., or Sarepta, drug EXONDYS 51®, also known as eteplirsen, an exon skipping therapy targeting the skipping of exon 51, that was granted accelerated approval by the FDA, and PTC Therapeutics' EMFLAZA®, also known as deflazacort, a corticosteroid, which is indicated for the treatment of DMD in patients five years of age and older. Corticosteroid therapy, including treatment with prednisone, is often prescribed to treat the inflammation underlying DMD and to delay loss of ambulation. In DMD patients, corticosteroids have demonstrated efficacy, which is believed to be driven by reductions in activated NF-kB. However, corticosteroids primarily act through another pathway called the glucocorticoid receptor-mediated pathway, and can cause significant complications including growth suppression, excessive weight gain, behavioral changes, reduction in bone strength and compromise of the immune system. Over time, corticosteroids induce chronic myopathy in many diseases through induction of muscle protein breakdown, which ultimately leads to muscle damage. DMD patients treated with corticosteroids typically show an initial improvement in measures of muscle function but then resume a progressive decline. Approximately half of DMD patients treated with steroids lose the ability to walk by age thirteen and the vast majority are in wheelchairs by age sixteen. DMD patients typically live until their mid-twenties, despite the availability of corticosteroids.
Additionally, there are several treatments for DMD that are approved or under review in the European Union or are expected to be under review by regulatory agencies in the near future.
Sarepta's EXONDYS 51 is under review by the European Medicines Agency, or EMA, with a decision anticipated in the first half of 2018. Santhera Pharmaceuticals, or Santhera, has filed a marketing authorization application with the EMA for Raxone®, also known as idebenone, for the treatment of DMD in patients with respiratory function decline and not taking concomitant glucocorticoids. In September 2017, Santhera received a negative opinion from the Committee for Medicinal Products for Human Use, or CHMP, of the EMA for its Type II extension application for Raxone in DMD. Santhera appealed the decision, and in January 2018 Santhera announced that the CHMP had maintained its negative opinion on Raxone's application in DMD. Santhera has announced that it intends to collect further data to strengthen Raxone's clinical data package before refiling the marketing authorization application with the EMA. PTC Therapeutics' ataluren is conditionally approved in the European Union and several other countries for treatment of nonsense mutation DMD, or nmDMD, under the trade name Translarna™. PTC Therapeutics received a Complete Response Letter, or CRL, from the FDA for ataluren's new drug application, or NDA, for the treatment of nmDMD in October 2017, and, in February 2018, the Office of New Drugs of the FDA reiterated the FDA's prior decision and denied PTC Therapeutics' appeal of the CRL. EXONDYS 51 and ataluren target mechanisms to increase levels of dystrophin in muscles. Each of these agents addresses a specific type of genetic mutation in order to produce a partially functional dystrophin protein. The therapeutic goal of these product candidates is to reduce disease severity and extend survival in those DMD patients who are candidates for therapy with these agents. Based on the prevalence of the specific mutations that EXONDYS 51 and ataluren are designed to address, they would be expected to be effective in an aggregate of approximately 26% of DMD patients. The company believe that DMD patients, including those treated with these dystrophin-targeted therapies, will continue to require treatments to reduce muscle inflammation and degeneration and enhance muscle regeneration.
Edasalonexent for the Treatment of Duchenne Muscular Dystrophy
Based on the mechanism of action by which edasalonexent suppresses NF-kB and the results that Catabasis Pharmaceuticals has seen in preclinical models of DMD, the company believe that edasalonexent has the potential to combine reduction of inflammation and muscle degeneration with positive effects on muscle regeneration, all of which may allow patients to retain muscle function longer. In addition, the company believe that edasalonexent has the potential to be an effective therapy in all DMD patients, regardless of the underlying mutation, and to provide significant benefit to patients, both as monotherapy and when used in combination with other therapies, including dystrophin-targeted therapies. If the company receive marketing approval, the company intend to commercialize edasalonexent in North America itself and commercialize edasalonexent outside of North America either itself or with a collaborator.
In September 2016, the company announced a pre-clinical joint research collaboration with Sarepta to explore a combination drug treatment approach for DMD. In the collaboration, increased dystrophin protein expression has been seen with an exon-skip modality in combination with edasalonexent in a mouse model of DMD.
Edasalonexent Clinical Development
MoveDMD Phase 1/2 Trial of Edasalonexent in Patients with DMD
The company's MoveDMD Phase 1/2 trial enrolled ambulatory boys ages four to seven with a genetically confirmed diagnosis of DMD who were steroid naive or had not used steroids for at least six months prior to the trial. Boys enrolled in the trial were not limited to any specific dystrophin mutations and the 31 boys in the trial had 26 different dystrophin mutations. The MoveDMD trial was designed to be conducted in three sequential parts, Phase 1, Phase 2, and an open-label extension, which is on-going.
In February 2018, the company reported observation of consistent improvements in all four assessments of muscle function in the open-label extension portion of the MoveDMD trial through more than a year of oral 100 mg/kg/day edasalonexent treatment compared to the rates of change in the control period for boys prior to receiving edasalonexent treatment: the three age-appropriate timed function tests (10-meter walk/run, 4-stair climb and time to stand), as well as the North Star Ambulatory Assessment, NSAA, an integrated global assessment of muscle function. At the time of the most recent open-label extension data analysis on functional assessments, all 13 boys continuing to participate had received 100 mg/kg/day of edasalonexent for 48 weeks and 8 had reached 60 weeks of 100 mg/kg/day treatment. Changes during the control period prior to boys receiving edasalonexent were measured for 16 boys who commenced edasalonexent 100 mg/kg/day treatment either at the beginning of Phase 2 or at the beginning of the open-label extension. The trial was not powered to detect statistically significant changes in any of the assessments of muscle function during Phase 2 or the open-label extension, and statistically significant changes were not detected in these measures.
Additional supportive measures of muscle health also reinforce the positive edasalonexent treatment effects observed in the 100 mg/kg/day treatment group. All four muscle enzymes tested (creatine kinase, alanine aminotransferase, aspartate aminotransferase and lactate dehydrogenase) were significantly decreased compared to baseline following edasalonexent treatment at 12 weeks and later time points through 60 weeks (p£0.05), consistent with the ability of edasalonexent to slow muscle degeneration and improve muscle integrity. Biomarker results showed that C-reactive protein, or CRP, was significantly decreased with edasalonexent at 12, 24, 36 and 48 weeks compared to baseline in the 100 mg/kg/day treatment group (p£0.001). CRP is a well-characterized blood test marker that provides a global assessment of inflammation and is elevated in boys affected by DMD. The significant decrease observed in CRP supports a conclusion that the biological activity of edasalonexent in inhibiting NF-kB can decrease inflammation.
Edasalonexent was well tolerated in the MoveDMD trial with no clinical safety signals observed to date. The majority of adverse events, or AEs, have been mild in nature with no serious AEs. The most common treatment-related AEs were gastrointestinal, primarily mild and transient diarrhea and, in Phase 2, vomiting. There were no treatment-related serious adverse events, no drug discontinuations and no dose reductions. Additionally, boys with DMD in this age range typically have resting tachycardia, a heart rate that exceeds the normal resting rate, and the heart rate of the boys treated with edasalonexent decreased toward age-normative values during treatment through the last measurements taken at 48 weeks.
In October 2017, the company reported results following 24 and 36 weeks of edasalonexent treatment from the open-label extension similar to results observed following 48 and 60 weeks of treatment. Consistent improvements were seen in all four assessments of muscle function after 24 and 36 weeks of oral 100 mg/kg/day edasalonexent treatment compared to the rates of change in the control period for boys prior to receiving edasalonexent treatment. Lower leg muscle magnetic resonance imaging, or MRI T2 rate of change was significantly improved in comparison to progression during the off-treatment control period (p<0.05). MRI, is a non-invasive imaging technique that allows investigators to view muscle structure and composition and measure disease status in children with DMD. Changes in MRI measures, particularly fat fraction, have been correlated in natural history studies with longer-term changes in clinically meaningful measures of functional activity. MRI T2 increases over time in DMD and is highly correlated with worsening timed function tests. All four muscle enzymes were significantly decreased compared to baseline following edasalonexent treatment at 12 weeks and later time points through 36 weeks (p<0.05). At the time of the first open-label extension data analysis, all 14 boys continuing to participate had received 100 mg/kg/day for 24 weeks and 11 had completed 36 weeks of 100 mg/kg/day edasalonexent treatment. The company transitioned all patients participating in open-label extension of the trial who were on the 67/mg/kg/day dose to the 100 mg/kg/day dose following availability of 12-week results, on average approximately after 36 weeks of treatment, and subsequently transitioned participating boys in the open-label extension to a higher dose of 133 mg/kg/day, given the safety profile the company had seen at that point, to provide additional information for the development of edasalonexent.
Thirty-one boys enrolled in Phase 2 of the MoveDMD trial and all completed the 12-week randomized, double-blind, placebo-controlled Phase 2 portion of the trial. Two dose levels of edasalonexent were evaluated, 67 mg/kg/day, dosed as 33 mg/kg in capsules twice a day, and 100 mg/kg/day, dosed as 33 mg/kg in capsules three times a day. In the Phase 2 portion of the trial, the company assessed the effects of edasalonexent using MRI T2 as an early biomarker at 12 weeks. Phase 2 of the MoveDMD trial was conducted at five sites in the United States. The company announced in January 2017 that the primary efficacy endpoint of average change from baseline to week 12 in the MRI T2 composite measure of lower leg muscles for the pooled edasalonexent treatment groups compared to placebo was not met (0.37 milliseconds for the pooled edasalonexent treatment groups and 0.138 milliseconds for the edasalonexent 100 mg/kg/day treatment group versus 0.47 milliseconds for placebo), although the company observed directionally positive results that were not statistically significant. A smaller increase in MRI T2 is believed to correlate with less muscle inflammation.
Results from the 12-week Phase 2 demonstrated that the edasalonexent 100 mg/kg/day treatment group consistently showed numerical improvement versus placebo in all assessments of muscle function. Similarly, the 67 mg/kg/day treatment group consistently showed numerical improvement versus placebo across multiple assessments of muscle function, although the changes were not statistically significant and mixed compared to the 100 mg/kg/day treatment group, consistent with an edasalonexent dose response. Additionally, edasalonexent plasma exposure in Phase 2 of the MoveDMD trial was consistent with that observed previously in Phase 1.
Phase 3 Clinical Trial
The company plan to initiate a single global Phase 3 trial in DMD in the first half of 2018 and expect to report top-line results from this trial in 2020, dependent on raising capital. The purpose of the trial is to evaluate the efficacy and safety of edasalonexent for registration purposes. The design of this randomized, double-blind, placebo-controlled trial has been informed by discussions with FDA and EMA. The Phase 3 trial is expected to have many key elements in common with the Phase 2 trial, including the patient population and functional endpoints. The company anticipate enrolling approximately 125 boys between their 4th and 7th birthday who have not been on steroids for at least 6 months. Boys on a stable dose of EXONDYS 51 may be eligible to enroll. The primary efficacy endpoint will be change in the North Star Ambulatory Assessment score after 12 months of treatment with edasalonexent compared to placebo. Key secondary endpoints are planned to include age-appropriate timed function tests. The company also expect to include assessments of growth, cardiac and bone health.
Completed Clinical Trials
To date, Catabasis Pharmaceuticals has studied edasalonexent in three completed Phase 1 clinical trials, in addition to Phase 1 of the MoveDMD trial, which is described above. The design for each of these other clinical trials are discussed below.
Edasalonexent—Completed Phase 1 Clinical Trials
| Trial | Description | Duration Of | Total | Treated with |
|---|---|---|---|---|
| Dosing | edasalonexent | |||
| CAT-1004-101 | Randomized, double-blind, placebo-controlled, single ascending dose clinical trial to evaluate safety, tolerability and pharmacokinetics of edasalonexent in healthy subjects | 1 day | 52 | 39 |
| CAT-1004-102 | Randomized, double-blind, placebo-controlled multiple ascending dose clinical trial to evaluate safety, tolerability, pharmacokinetics and pharmacodynamics of edasalonexent in adults with Type 2 diabetes | 14 days | 44 | 32 |
| CAT-1004-103 | Single-blind biomarker trial in healthy adults to compare activity of edasalonexent, a combination of salicylate and DHA, or placebo on activated NF-kB | 1 day | 9 | 8 |
Phase 1 Single Ascending Dose Trial (CAT-1004-101): The company conducted a randomized, double-blind, placebo-controlled, single ascending dose Phase 1 clinical trial in 52 healthy volunteers at a single site in the United States to assess the safety, tolerability and pharmacokinetics of edasalonexent in both fasted and fed states. The participants were randomized to receive edasalonexent or placebo. Edasalonexent was administered orally in soft gelatin capsules at doses ranging from 300 mg to 6000 mg and appeared to be well tolerated. The most common adverse events in the fasted state were headache, diarrhea and dizziness, and the majority of the adverse events in the fasted state were mild in severity. The most common adverse events in the fed state were diarrhea, headache and abdominal pain, and all of the adverse events in the fed state were mild in severity. No treatment-related severe adverse events were reported. There were no observed trends in laboratory, vital signs or electrocardiogram results following edasalonexent administration in either the fasted or fed state.
Phase 1 Multiple Ascending Dose Trial (CAT-1004-102): The company conducted a randomized, double-blind, placebo-controlled, multiple ascending dose Phase 1 clinical trial in 44 subjects at a single center in the United States to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of edasalonexent. These subjects had Type 2 diabetes and mild background inflammation, which enabled it to assess the activity of edasalonexent on activated NF-kB. Subjects were randomized to receive edasalonexent or placebo. Edasalonexent was administered orally in soft gelatin capsules at total daily doses ranging from 300 mg to 4000 mg and appeared to be well tolerated. The majority of the adverse events were mild in severity. No treatment-related severe adverse events were reported.
Edasalonexent was rapidly absorbed in plasma, with mean maximum and overall plasma exposure generally increasing with escalating single or multiple doses of edasalonexent. Neither component bioactive, salicylate or DHA, was detected in plasma at levels above background, again consistent with intracellular cleavage of edasalonexent and intracellular delivery of the component bioactives.
The company also observed that edasalonexent inhibited activated NF-kB by two methods. For the first method, the company stimulated NF-kB activity ex vivo in whole blood from subjects treated with edasalonexent or placebo, and then observed NF-kB activity in isolated monocytes. NF-kB activity was reduced in a majority of subjects following two weeks of edasalonexent treatment but not following treatment with placebo. For the second method, the company performed gene expression analyses on whole blood taken from subjects prior to treatment and after two weeks of treatment with edasalonexent or placebo. Edasalonexent, but not placebo, significantly reduced the expression of a set of genes that are controlled by NF-kB.
Phase 1 NF-kB Biomarker Trial (CAT-1004-103): The company conducted a single-blind, crossover Phase 1 clinical trial with edasalonexent in nine healthy adult volunteers at a single center in the United States to compare activity of a single dose of 2000 mg of edasalonexent on activated NF-kB to a combination of salicylate and DHA or placebo. No adverse events were reported in this clinical trial. The salicylate and DHA were dosed at approximately equivalent amounts to those contained in the edasalonexent conjugate. The company assessed NF-kB activity in peripheral blood mononuclear cells isolated from subjects before dosing and two hours after dosing. Prior to the determination of NF-kB activity, the company stimulated whole blood with lipopolysaccharide, or LPS, to activate the NF-kB pathway. Treatment of subjects with edasalonexent significantly reduced the level of activated NF-kB, as measured by nuclear p65, a surrogate marker for activated NF-kB. In contrast, no change in the level of activated NF-kB was observed upon treatment with the combination of salicylate and DHA, or upon treatment with placebo. In this trial, edasalonexent, which is a SMART Linker conjugate of salicylate and DHA, exhibited greater activity on the NF-kB pathway than the combination of its component bioactives.
Edasalonexent Orphan Drug, Fast Track and Rare Pediatric Disease Designations
The FDA has granted edasalonexent orphan drug, fast track and rare pediatric disease designations for the treatment of DMD. A product may be designated by the FDA as an "orphan drug" if it is intended to treat a rare disease or condition affecting fewer than 200,000 individuals in the United States. If a product with orphan status receives the first FDA approval for the disease or condition for which it has such designation, the FDA will not approve another sponsor's marketing application for the same product for the same use or indication before the expiration of seven years, except in certain limited circumstances. The FDA fast track process is designed to expedite the development and review of drugs to treat serious or life-threatening conditions and demonstrate the potential to address unmet medical needs. Companies that receive fast track designation are allowed to submit NDAs on a rolling basis, expediting the FDA review process, and benefit from more frequent communication with the FDA to discuss all aspects of clinical development. In addition, drugs that receive fast track designation are eligible for accelerated approval and priority review if certain criteria are met. The FDA's rare pediatric disease designation gives it the potential to receive a priority review voucher if edasalonexent is approved. However, the rare pediatric disease program is set to expire in September 2020.
The EC has granted orphan medicinal product designation to edasalonexent for the treatment of DMD. Similar to the FDA orphan drug designation, the EC may designate a product as an orphan medicinal product if it is intended for the treatment of a life-threatening or chronically debilitating condition affecting not more than five in ten thousand persons. In Europe, marketing authorization for an orphan medicinal product generally leads to up to a ten-year period of market exclusivity if the product candidate is granted marketing authorization in the European Union.
Edasalonexent Expansion Indication Opportunities
In addition to its work in DMD, Catabasis Pharmaceuticals is evaluating other diseases where the inhibition of NF-kB may be beneficial for further therapeutic applications of edasalonexent. There are a number of other rare diseases where NF-kB is believed to play an important role, such as Becker muscular dystrophy, which is a type of muscular dystrophy and is characterized by slowly progressive muscle weakness of the legs and pelvis, and IgA nephropathy, a kidney disease that is believed to result from activation of mucosal immunity leading to the synthesis of aberrantly glycosylated polymeric immunoglobulin A1, which enters the circulation and lodges in a patient's kidneys interfering with their proper function.
CAT-5571
CAT-5571 is a SMART Linker conjugate that contains cysteamine, a naturally occurring molecule that is a degradation product of the amino acid cysteine, and DHA. Catabasis Pharmaceuticals is developing CAT-5571 initially as a potential oral treatment for CF, designed to restore host defense by activating autophagy. Autophagy is a mechanism for recycling cellular components and digesting pathogens, which is depressed in CF. People with CF suffer from persistent lung infections from opportunistic pathogens such as P. aeruginosa and B. cenocepacia, causing chronic infections that are difficult to eradicate and ultimately lead to respiratory failure.
CAT-5571 has been shown to restore autophagy, reestablish host defense and enhance the clearance of pathogens, including P. aeruginosa and B. cenocepacia in preclinical models of CF. The company plan to complete investigational new drug, or IND, application-enabling activities for CAT-5571 in 2018 and to advance CAT-5571 into a Phase 1 clinical trial in the second half 2018 and report top-line results in 2019, based on its current operating plan.
Cystic Fibrosis
CF is a rare, chronic, genetic, life-shortening orphan disease that affects over 70,000 patients worldwide, predominantly in the Caucasian population. In CF, a malfunctioning cystic fibrosis transmembrane conductance regulator ion channel impairs chloride secretion, with deleterious effects on multiple organs, and particularly devastating effects on pulmonary, intestinal and pancreatic function. Patients affected with CF are also predisposed to respiratory failure caused by persistent lung infections, notably bacteria and most commonly P. aeruginosa, that are difficult to treat with standard antibiotics. CF patients have frequent pulmonary exacerbations due to their inability to clear the persistent lung infections. Advancement in research and treatments have extended the life expectancy for those living with CF, however, there is currently no cure.
CAT-4001
CAT-4001 is a SMART Linker conjugate that the company designed to combine the potentially beneficial activities of monomethyl fumarate and DHA on the Nrf2 and NF-kB pathways. CAT-4001 is a small molecule designed to activate the Nrf2 pathway and inhibit the NF-kB pathway. Catabasis Pharmaceuticals is developing CAT-4001 initially for the treatment of severe, rare neurodegenerative diseases, such as FA and ALS, two diseases of the central nervous system in which the Nrf2 and NF-kB pathways have been implicated, irrespective of mutation status. Nrf2 is a gene transcription factor, a protein that works inside of cells to control the expression of genes, that control the body's response to cellular stress and oxidative damage. Catabasis Pharmaceuticals is conducting preclinical activities with CAT-4001.
Friedreich's Ataxia
Friedreich's ataxia is a rare genetic disease that causes nervous system damage and compromises motor coordination. FA is caused by a defect in the frataxin gene, which regulates iron levels in the mitochondria. In the majority of cases, the genetic defect in FA causes a reduction in the production of the frataxin protein and iron levels in mitochondria become poorly regulated. Progressive degeneration of central and peripheral nervous systems in FA patients causes impaired gait and coordination, muscle loss and fatigue. Disease progression varies, but generally, the patient is confined to a wheelchair within 10 to 20 years after the appearance of the first symptoms. Patients may become completely incapacitated in later stages of the disease.
FA occurs in both males and females and is estimated to affect 1 in 50,000 individuals. Based on this prevalence rate, the company believe there are up to 6,000 patients with FA in the US and up to 15,000 FA patients in the European Union.
The Friedreich's Ataxia Research Alliance announced in January 2016 that the company were the recipient of the Kyle Bryant Translational Research Award. The Kyle Bryant Translational Research Award specifically focuses on pre-clinical and clinical investigations that target treatments for FA.
Amyotrophic Lateral Sclerosis
ALS, sometimes called Lou Gehrig's disease or classical motor neuron disease, is a rapidly progressive, fatal neurological disease that attacks the nerve cells responsible for controlling voluntary muscles. Eventually, muscle weakness and atrophy occur. People with ALS lose the ability to stand and walk, and use their hands and arms. In later stages of the disease, individuals have difficulty breathing as the muscles of the respiratory system weaken. Although ventilation support can enable breathing and prolong survival, it does not affect the progression of ALS. Most people with ALS die from respiratory failure, usually within three to five years of diagnosis.
According to the ALS Association, approximately 5,600 people in the United States are diagnosed with ALS each year. The incidence of ALS is two per 100,000 people, and it is estimated that as many as 30,000 Americans may have the disease at any given time.
CAT-2000 Series
The company's CAT-2000 compounds are SMART Linker conjugates of nicotinic acid and EPA. The linkers for its CAT-2000 series compounds are cleaved through intracellular enzymatic hydrolysis, to release the component bioactives to inhibit the Sterol Regulatory Element Binding Protein, or SREBP, pathway which has been shown to be important in the development of nonalcoholic steatohepatitis, or NASH. In vivo, CAT-2000 molecules have demonstrated efficacy in multiple preclinical models of NASH. The company believe that its portfolio of CAT-2000 molecules provides an opportunity to develop a therapy for NASH. The company intend to pursue a partnership for further development of the CAT-2000 series in NASH.
Sales and Marketing
Given its stage of development, Catabasis Pharmaceuticals has not yet established a commercial organization or distribution capabilities, nor have the company entered into any collaboration or co-promotion arrangements. If Catabasis Pharmaceuticals is able to progress its edasalonexent program, the company intend to commercialize edasalonexent in North America itself and commercialize edasalonexent outside of North America either itself or with a collaborator. In addition, the company intend to expand the drug development applications of its SMART Linker drug discovery platform through selective collaborations with leading biotechnology and pharmaceutical companies.
Manufacturing and Supply
Each of its SMART Linker conjugate product candidates is a small molecule compound manufactured from component raw materials. The omega-3 fatty acid materials that the company use as bioactives are purified from natural sources by established pharmaceutical fine chemicals manufacturers. The other bioactive and linker raw materials that the company use are also readily available from established pharmaceutical intermediate manufacturers. The components are conjugated to form the SMART Linker product candidate using well understood, conventional chemistries.
The company currently have no manufacturing facilities and limited personnel with manufacturing experience. The company rely on contract manufacturers to produce both drug substance and drug product required for its clinical trials. The company plan to continue to rely upon contract manufacturers and, potentially, collaborators to manufacture commercial quantities of its products, if approved.
Competition
The development and commercialization of new drugs is highly competitive. If the company successfully develop and commercialize any of its product candidates, the company and any future collaborators will face competition from pharmaceutical and biotechnology companies worldwide. Many of the entities developing and marketing potentially competing products have significantly greater financial resources and expertise than the company do in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing. The company's commercial opportunity will be reduced or eliminated if its competitors develop and commercialize products that are more effective, have fewer side effects, are more convenient or are less expensive than any products that the company may develop.
The key competitive factors affecting the success of its product candidates, if approved, are likely to be their efficacy, safety, convenience, price and the availability of coverage and reimbursement from government and other third-party payors.
Edasalonexent for Duchenne Muscular Dystrophy
There are currently only two therapies approved in the United States for the treatment of DMD. Sarepta's drug EXONDYS 51 was approved by the FDA for the treatment of DMD under the accelerated approval pathway in September 2016 for patients who have a confirmed mutation of the DMD gene that is amenable to exon 51 skipping. In addition, in February 2017, the FDA granted approval of PTC Therapeutics' drug EMFLAZA™, for the treatment of DMD in patients five years and older. Outside of the United States, PTC Therapeutics' drug ataluren, also known as Translarna™, has been conditionally approved within the European Union Member States, Iceland, Liechtenstein, Norway, Israel and South Korea for the treatment of nmDMD. Although not previously approved for the treatment of DMD, corticosteroid therapy, including prednisone, is considered standard of care and is often prescribed to treat the inflammation underlying DMD and to delay loss of ambulation.
A number of companies are developing therapies to treat DMD in patients with specific mutations in the dystrophin gene. In addition to EXONDYS 51, Sarepta has two additional exon-skipping therapies for DMD in Phase 3 clinical development. These agents, SRP-4053 and SRP-4045, target skipping of exons 53 and 45, respectively. Daiichi-Sankyo is developing an exon-skipping product candidate for DMD patients with out-of-frame deletion mutations amenable to exon 45 skipping, and announced in February 2016 that it began its first Phase 1/2 clinical trial for its product candidate, DS-5141b, in Japan. NS Pharma has a compound, NS-065/NCNP-01, in Phase 2 clinical development in the United States and Japan for patients with mutations amenable to exon 53 skipping. NS-065/NCNP-01 received orphan drug designation in the US and was granted Fast Track designation by the FDA. Based on the prevalence of the specific mutations that these product candidates being developed by Sarepta, Daiichi-Sankyo and NS-Pharma are designed to address, they would be expected to have the potential to be effective in an aggregate of approximately 16% of DMD patients. In addition, Wave Life Sciences Ltd. initiated a Phase 1 clinical trial in DMD in November 2017 for its exon 51 skipping candidate, WVE-210201.
In addition to exon-skipping therapies, other companies have alternative therapeutic approaches to the treatment of DMD in late stage clinical development. As described further above under "—Unaddressed Market Opportunity", several companies with alternative therapeutic approaches to DMD, including Santhera and PTC Therapeutics, have had delays in obtaining the necessary approvals from United States and European regulatory agencies. In addition, other alternative therapeutic approaches are in later stage clinical trials, including Italfarmaco S.p.A.'s Phase 3 trial in ambulant DMD boys for givinostat, a histone deacetylase inhibitor. Each of Pfizer, Inc., or Pfizer, and F. Hoffman-La Roche Ltd., or Roche, have anti-myostatin inhibitors in Phase 2/3 clinical trials. A number of companies also have products candidates in earlier clinical development for DMD, including Akashi Therapeutics, Astellas, Capricor Therapeutics, Cardero Therapeutics, EspeRare, Fibrogen, Phrixus Pharmaceuticals, Reveragen, Summit Plc, and Taiho Pharmaceuticals. If successfully developed, some of these alternative therapeutic approaches may be applicable to all DMD patients regardless of underlying mutation status. Additionally, several gene therapy programs targeting the dystrophin gene have entered clinical development, including Solid Biosciences SGT-001, Pfizer's PF-06939926, and Sarepta's GALGT2 and micro-dystrophin programs being conducted in collaboration with Nationwide Children's Hospital.
Intellectual Property
The company strive to protect the proprietary technologies that the company believe are important to its business, including pursuing and maintaining patent protection intended to cover the composition of matter of its product candidates, their methods of use, related technologies and other inventions that are important to its business. In addition to patent protection, the company also rely on trade secrets to protect aspects of its business that are not amenable to, or that the company do not consider appropriate for, patent protection, including certain aspects of its SMART Linker drug discovery platform.
The company's commercial success depends in part upon its ability to obtain and maintain patent and other proprietary protection for commercially important technologies, inventions and know-how related to its business, defend and enforce its intellectual property rights, in particular, its patent rights, preserve the confidentiality of its trade secrets and operate without infringing valid and enforceable intellectual property rights of others.
The patent positions for biotechnology and pharmaceutical companies like it are generally uncertain and can involve complex legal, scientific and factual issues. In addition, the coverage claimed in a patent application can be significantly reduced before a patent is issued, and its scope can be reinterpreted and even challenged after issuance. As a result, the company cannot guarantee that any of its product candidates will be protected or remain protectable by enforceable patents. The company cannot predict whether the patent applications Catabasis Pharmaceuticals is currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient proprietary protection from competitors. Any patents that the company hold may be challenged, circumvented or invalidated by third parties.
As of December 31, 2017, its patent estate included over 20 issued U.S. patents, over 100 issued foreign patents, over 10 pending U.S. patent applications and over 35 pending foreign patent applications.
With regard to edasalonexent, Catabasis Pharmaceuticals has six issued U.S. patents with composition of matter and method of use claims covering edasalonexent and its use. The issued U.S. patents are expected to expire in 2029, without taking a potential patent term extension into account. In addition, Catabasis Pharmaceuticals has patents that have been granted in various countries and regions including Australia, China, Europe, Israel, India, Japan, Korea, Mexico and New Zealand, which are expected to expire in 2029, without taking potential patent term extensions into account, and at least five pending patent applications in various other countries and regions in North America, South America, and Asia, which, if issued, are expected to expire in 2029, without taking potential patent term extensions into account.
With regard to CAT-5571, Catabasis Pharmaceuticals has four issued U.S. patents with composition of matter and method of use claims covering CAT-5571 and its use, which are scheduled to expire in 2030, without taking potential patent term extensions into account. The company also have over 10 patent applications pending in the U.S. and in other regions including North America, South America, Europe and Asia with claims covering CAT-5571 and related compounds and their use, including their use in the treatment of cystic fibrosis.
With regard to CAT-4001, Catabasis Pharmaceuticals has two issued U.S. patents with composition of matter and method of use claims covering CAT-4001 and its use. These U.S. patent are scheduled to expire in 2031, without taking a potential patent term extension into account. In addition, Catabasis Pharmaceuticals has patents that have been granted in various countries and regions including Australia, China, Europe, Israel, Japan, Mexico and New Zealand, which are expected to expire in 2031, without taking potential patent term extensions into account, and at least 5 pending patent applications in various other countries and regions in North America, South America, and Asia, which, if issued, are expected to expire in 2031, without taking potential patent term extensions into account.
With regard to CAT-2003 and CAT-2054, Catabasis Pharmaceuticals has six issued U.S. patents with composition of matter and method of use claims covering CAT-2003 and CAT-2054 and their use. These U.S. patents are scheduled to expire in 2030 and 2031, without taking potential patent term extensions into account. In addition, Catabasis Pharmaceuticals has patents that have been granted in several different countries and regions including Australia, China, Europe, Israel, Japan, Korea, Mexico and New Zealand, which are expected to expire in 2030, without taking potential patent term extensions into account and four pending applications in various other countries and regions including North and South America, and Asia, which, if issued, are expected to expire in 2030, without taking patent term extensions into account.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which the company file, the patent term is 20 years from the earliest date of filing a non-provisional patent application.
In the United States, the term of a patent covering an FDA-approved drug may be eligible for a patent term extension under the Hatch-Waxman Act as compensation for the loss of patent term during the FDA regulatory review process. The period of extension may be up to five years beyond the expiration of the patent, but cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval. Only one patent among those eligible for an extension may be extended. Similar provisions are available in Europe and in certain other jurisdictions to extend the term of a patent that covers an approved drug. It is possible that issued U.S. patents covering edasalonexent, CAT-5571, CAT-4001, CAT-2003 and CAT-2054 may be entitled to patent term extensions. If its product candidates receive FDA approval, the company intend to apply for patent term extensions, if available, to extend the term of patents that cover the approved product candidates. The company also intend to seek patent term extensions in any jurisdictions where they are available, however, there is no guarantee that the applicable authorities, including the FDA, will agree with its assessment of whether such extensions should be granted, and even if granted, the length of such extensions.
In addition to patent protection, the company also rely on trade secret protection for its proprietary information that is not amenable to, or that the company do not consider appropriate for, patent protection, including, for example, certain aspects of its manufacturing processes and conjugate selection methodologies. However, trade secrets can be difficult to protect. Although the company take steps to protect its proprietary information, including restricting access to its premises and its confidential information, as well as entering into agreements with its employees, consultants, advisors and potential collaborators, third parties may independently develop the same or similar proprietary information or may otherwise gain access to its proprietary information. As a result, the company may be unable to meaningfully protect its trade secrets and proprietary information.
Research and Development Expenses
For the years ended December 31, 2017, 2016 and 2015, the company incurred approximately $18.7 million, $25.5 million, and $23.0 million, respectively, on research and development activities.




