Galena Biopharma
Overview
Galena Biopharma (GALE) is a clinical-stage biopharmaceutical company focused on novel cancer immunotherapeutics for a broad range of cancer indications. The company's lead product candidate, galinpepimut-S, or GPS, is a cancer i unotherapeutic agent licensed from Memorial Sloan Kettering Cancer Center, or MSK, that targets the Wilms tumor 1, or WT1, protein, which is present in 20 or more cancer types. Based on its mechanism of action as a directly immunizing agent, GPS has the potential as a monotherapy or in combination with other immunotherapeutic agents to address a broad spectrum of hematologic, or blood, cancers and solid tumor indications. Phase 2 clinical trials for GPS have been completed and Galena Biopharma has planned Phase 3 clinical trials (pending funding availability) for two indications, acute myeloid leukemia, or AML, and malignant pleural mesothelioma, or MPM. GPS is also in development as a potential treatment for multiple myeloma, or MM, and ovarian cancer. The company plan to study GPS in up to four additional indications: as a combination therapy in small cell lung cancer, colorectal cancer, triple-negative breast cancer; and, as a monotherapy in chronic myelogenous leukemia, or CML. The company received Orphan Drug Product Designations from the U.S. Food and Drug Administration, or FDA as well as Orphan Medicinal Product Designations from the European Medicines Agency, or EMA, for GPS in AML and MPM, as well as Fast Track Designation for AML and MPM from the FDA.
The company's pipeline also includes the legacy development programs of its pre-Merger company, including novel cancer immunotherapy programs for NeuVax™ (nelipepimut-S; a vaccine against the E75 peptide derived from the human epidermal growth factor 2 -or HER2- protein), GALE-301 (a vaccine against the E39 peptide derived from the folate binding protein, or FBP) and GALE-302 (a vaccine against the J65 peptide derived from FBP) and GALE-401 (a controlled release version of the approved drug anagrelide). NeuVax is currently in multiple investigator-sponsored Phase 2 clinical trials in breast cancer, including a prospective, randomized, single-blinded, controlled Phase 2b independent investigator-sponsored clinical trial of trastuzumab (Herceptin®) +/- NeuVax in HER2 1+/2+ breast cancer patients in the adjuvant setting to prevent recurrences. On April 2, 2018, the company announced that a pre-specified interim analysis, conducted by an independent Data Safety Monitoring Board, or DSMB, of the efficacy and safety data for the study demonstrated a clinically meaningful difference in median disease-free survival, or DFS, in favor of the active arm (NeuVax + Herceptin), a primary endpoint of the study. Based on these results, and the DSMB’s recommendation, the company plan to expeditiously seek regulatory guidance by the FDA for further development of the combination of NeuVax + Herceptin in Triple Negative Breast Cancer, or TNBC, considering the statistically significant benefit of the combination therapy seen in this population with large unmet medical need.
GALE-301 and GALE-302 have completed early stage trials in ovarian, endometrial and breast cancers. GALE-401 is being developed for the treatment of elevated platelets in patients with myeloproliferative neoplasms, or MPNs, and a completed Phase 2 clinical trial in patients with essential thrombocythemia, or ET for this clinical candidate. Since the closing of the Merger, management has been evaluating GALE-301, GALE-302, and GALE-401 for potential internal development, strategic partnership, or other types of product rationalizations.
Strategy
The company seek to use its expertise and understanding of cancer immunotherapy and general cancer therapeutic product development to develop novel products that have the potential to transform the treatment of cancer patients. The key components of its strategy are as follows:
- Continue to rapidly advance its first-in-class cancer immunotherapy product candidates and other new products through clinical development. The company intend to continue to execute a focused clinical development plan that takes its product candidates through approval by regulatory authorities. This includes developing GPS as both a monotherapy or in combination, in addition to exploring opportunities for the other product candidates in its pipeline. The entire GPS clinical program currently targets up to eight tumor types, namely AML, MPM, MM, ovarian cancer, small cell lung cancer, colorectal cancer, triple negative breast cancer, and CML. The company may pursue additional development of GPS for other indications, both as a monotherapy or in combination with other therapeutic agents.
- GPS monotherapy: GPS has completed Phase 2 clinical trials and has Phase 3 clinical trials planned (pending funding availability) for AML and MPM. There is also an ongoing Phase 2 clinical trial of GPS for MM as monotherapy. The company also have plans to pursue additional clinical development programs for GPS as a monotherapy, including in chronic myelogenous leukemia, or CML, and AML treated with hypomethylators.
- GPS combination therapy: GPS has an ongoing Phase 1/2 clinical trial for ovarian cancer, in combination with nivolumab (Opdivo®) (the clinical trial is independently sponsored by MSK). The company plan to test GPS in combination with other therapeutic agents for various solid and hematologic cancers. The company's leading combination clinical program will be in collaboration with a Merck & Co., Inc., Kenilworth, N.J., USA subsidiary (known as MSD outside the United States and Canada), or Merck subsidiary. The purpose of the trials is to determine if the administration of GPS in combination with the PD1 blocker pembrolizumab (Keytruda®) has the potential to demonstrate clinical activity in the presence of macroscopic disease, where monotherapy with either agent would have a more limited effect.
- Utilize rare disease development pathways at the FDA and comparable foreign regulatory agencies to accelerate progression to late-stage development and early approval. A component of its strategy is to focus on rare types of cancers where its cancer immunotherapy product candidates may produce clinical benefit and where the company can take advantage of regulatory programs intended to expedite drug development in these types of rare cancers. The company received Orphan Drug Product Designation from the FDA as well as Orphan Medicinal Product Designation from the EMA for GPS in AML and MPM, as well as Fast Track Designation from the FDA for AML, MPM, and NeuVax. The company plan to apply for Orphan Drug Designation, Fast Track Designation, Breakthrough Therapy Designation and Priority Review from the FDA as well as Orphan Medicinal Product Designation, Priority Medicines Designations, and Conditional Authorizations from the EMA for any given indication, if applicable when pertinent data becomes available, to potentially reduce clinical trial expense and increase speed to commercialization.
- Enter into collaboration and license agreements with other biotechnology and pharmaceutical companies to develop its current product candidates and other future product candidates. WE seek out collaborations for additional opportunities and development of programs in its pipeline that require larger clinical trials or extensive commercial infrastructure. Specifically, the company plan to advance the development of NeuVax through a partnership or other strategic collaboration. Galena Biopharma is also evaluating licensing and other strategic options for GALE-301, GALE-302 and GALE-401.
- Selectively build focused commercial capabilities and establish commercial collaborations to maximize the value of its clinical development pipeline. Galena Biopharma has not yet defined its sales, marketing or product distribution strategy for GPS or any future product candidates. The company's future commercial strategy may include the use of strategic alliances, distributors, a contract sales force, or the establishment of its own commercial and specialty sales force to maximize the value of its pipeline.
The chart below summarizes the current status of its clinical development pipeline:
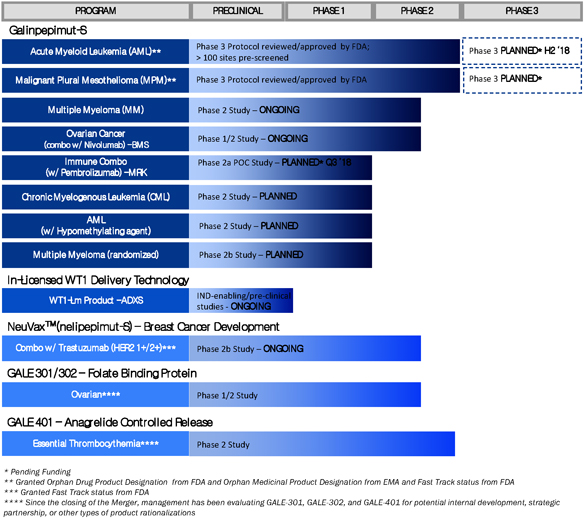
The Cancer Immunotherapy Industry
Overview
The principle behind cancer immunotherapy involves stimulating a person’s own immune system to selectively attack cancer cells while keeping normal cells unaffected or delivering certain immune system components in order to inhibit the spread of cancer. Cancer immunotherapy drugs now constitute a new mode of cancer treatment, alongside more established options such as surgery, chemotherapy, targeted therapy and radiation therapy. A July 2016 report by Kelly Scientific Publications estimates that immunotherapies may eventually be used in as many as 60% of cases of advanced cancer; further, based on a recent Allied Market Research report on the estimated entire market value of oncology drugs in 2020, cancer immunotherapies could represent up to 71% of that total value. Either in mono or in combination therapies, immunotherapies may produce long-term remissions or even operational “cures” for cancers that have been uniformly fatal until recently. Thus, cancer immunotherapy is an important and rapidly emerging field, which has led to exciting new clinical research studies and garnered the attention of investors, biotechnology and pharmaceutical companies, regulatory agencies, payors and hospital systems, cancer patients and their families and the general public at large.
Market
The global market for cancer drugs (including immunotherapy drugs) is expected to reach $161.3 billion by end of 2021, growing at a compound annual growth rate, or CAGR of around 7.4% between 2016 and 2021 (according to a December 2016 report by Zion Market Research). According to a September 2016 report by MarketsandMarkets, the global cancer immunotherapy market is expected to reach $119.4 billion by 2021 from $61.97 billion in 2016 at an estimated CAGR of 14.0%. The company estimate that by 2021, 74% of the oncology market worldwide will be supported by usage of cancer immunotherapies.
The first category of immunotherapies, immune synapse modulators (which includes checkpoint inhibitors and immune synapse co-stimulators), is likely to reach and exceed 90% of the immunotherapy market in the coming years, which leaves approximately 10% for the other three major categories, which include peptide cancer active immunizers such as its product candidate, GPS.
GPS targets malignancies and tumors characterized by an overexpression of the WT1 protein. The WT1 protein is one of the most widely expressed cancer proteins in multiple malignancies. A 2009 pilot project regarding the prioritization of cancer antigens conducted by the National Cancer Institute, or NCI, a division of the National Institutes of Health, or NIH, ranked the WT1 protein as a top priority for immunotherapy. WT1 is a protein that resides in the cell’s nucleus and participates in the process of cancer formation and progression. As such, it is classified as an “oncogene.” WT1 plays a key role in the development of the kidneys in fetal life, but then almost disappears from normal organs and tissues. In a wide variety of cancers (20 or more cancer types), WT1 becomes detectable again in the cells of these cancers. WT1 appears in large amounts (i.e., becomes “overexpressed”) in numerous hematological malignancies, including AML, MM and chronic myeloid leukemia, as well as in many solid malignancies such as MPM, gastrointestinal cancers (such as colorectal cancer), glioblastoma multiforme, triple-negative breast cancer, ovarian cancer and small-cell lung cancer. Overall, WT1 is expressed in at least 50% of tumor pathology specimens in 20 or more cancer types. The following figure shows the ratio of samples testing positive for WT1 to those testing negative for WT1 in a number of different malignancies.
WT1 EXPRESSION FREQUENCY ACROSS VARIOUS CANCERS
(Positive samples / Total samples)
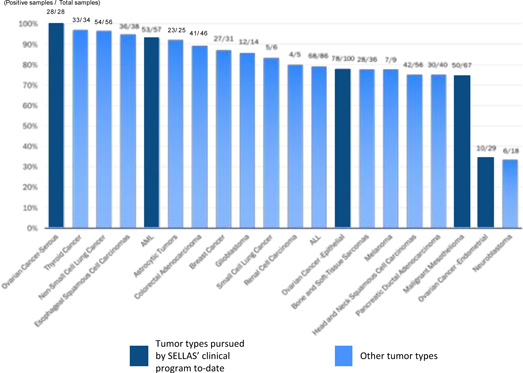
Data sampling overview from multiple studies in human tumor samples or cancer cell lines
The company's other cancer immunotherapy product, NeuVax (nelipepimut-S), utilizes a targeted approach based upon two key areas: preventing secondary recurrence of human epidermal growth factor receptor, or HER2, positive breast cancer because the number of breast cancer survivors continues to grow; and, primary prevention intended to prevent ductal carcinoma in situ, or DCIS, from becoming invasive breast cancer. Once a patient’s tumor becomes metastatic, the outcome is often fatal, making the prevention of recurrence a potentially critical component of overall patient care. The company's secondary recurrence programs for NeuVax primarily target patients in the adjuvant, or after-surgery setting who have relatively healthy immune systems but may still have residual disease. Minimal residual disease, or micrometastasis, that are undetectable by current radiographic scanning technologies, can result in breast cancer recurrence.
While GPS and NeuVax are both anti-cancer vaccines, they have some distinguishing features. GPS is tetravalent and Neuvax is monovalent. GPS is a direct immunogen emulsified into the clinically safe adjuvant Montanide, and administered subcutaneously after priming the immune system with recombinant human granulocyte macrophage-colony stimulating factor, or GM-CSF, Sargramostim. NeuVax, on the other hand, uses an immunodominant HER2 peptide combined with GM-CSF as the immune adjuvant, and is administered intradermally. Both GPS and NeuVax, however, work by harnessing the patient’s own immune system to seek out and attack any residual cancer cells. The company believe using peptide immunogens has many potential clinical advantages, including a favorable safety profile, because these therapies may lack the toxicities typical of most cancer therapies. Peptide immunogens also have the potential to induce immunologic memory and provide long-lasting protection with convenient modes of delivery.
Galinpepimut-S
Overview
GPS is a WT1-targeting peptide-based cancer immunotherapeutic being developed as a monotherapy and in combination with other therapeutic agents to treat different types of cancers that result from uninhibited tumor cell growth.
Cancer immunotherapy is an approach to cancer treatment that harnesses the body’s natural immune system response to fight and/or prevent such tumor growth. An essential feature of the immune system is its ability to recognize foreign, or non-self, threats, including cancerous growths, as distinct from normal, or self, cells. Despite originating from normal cells, tumor cells can be recognized as non-self because of their capacity to elicit the production of tumor antigens. These antigens may be released in the interstitial tissues and, eventually, the bloodstream or remain on the surface of cognate cancer cells. Such tumor-associated antigens, or TAAs, have been identified in most human cancers. The WT1 protein is one of the most widely expressed TAAs in multiple malignances.
The immune system is a network of tissues, cells, and signaling molecules that work to protect the body by recognizing and attacking foreign cells, including cancer cells. Several different types of cells are important for the development and maintenance of an immune response against cancer. The most crucial types of cells are antigen-presenting cells, or APCs, and lymphocytes. APCs include various subtypes, such as dendritic cells, monocytes and macrophages. Once a patient is exposed to a TAA (either by the presence of cancer itself or through active immunization through a vaccine type immunotherapeutic), this antigen gets recognized by the APC and becomes “processed” through digestion into smaller fragments within the APC. Subsequently, the APC “communicates” with a specific type of lymphocytes called T-cells. Inactive T-cells search for TAAs by transiently binding to antigens presented by major histocompatibility complexes, or MHCs, on the APCs. Notably, there is great variability in the expression of different subtypes of MHCs in the human population. The MHC system expresses the so-called human leukocyte antigens, or HLAs, and there are dozens of subclasses that determine the vigor and duration of any given T-cell response to a cancer among different patients. Consequently, active immunizers that work across many HLA types, such as GPS, are predicted to be more efficacious across larger segments of patient populations as compared to agents that act in the context of only one or few HLA types.
T-cells themselves also come in many variants. CD8 cells recognize the processed TAA fragment as foreign and respond. The CD8 cells also develop properties that can directly kill the TAA-expressing cancer cell by becoming “cytotoxic” CD8 cells. The CD8 cells, as well as the APCs, also activate CD4 cells, which are very important for the development of immunologic memory. Immunologic memory is developed when a host keeps a long-term trace of the TAA associated with the cancer and is a desirable result, as it allows the host to continue attacking the TAA associated with the cancer. Therefore, activation of CD4 cells helps avoid or mitigate immune “tolerance.” Immune tolerance is an undesirable result, as it dampens the host’s immune response against the cancer. This cascade of events is collectively called “cellular immunity” and is very important for anti-cancer activity of immunotherapeutic compounds such as GPS. Of note, once T-cells are activated, another class of lymphocytes, called B-cells, are also secondarily activated. B-cells are responsible for making antibodies against TAAs. These antibodies become expressed on the surface of the B-cells and are eventually secreted as soluble proteins in tissue fluids and blood. Such anti-cancer antibodies can be detected and have variable degree of activity against the cancer itself. This type of immunity is called “humoral immunity” and complements the actions and effects of the cellular immunity.
Key Features
GPS is a multi-peptide product that Galena Biopharma has exclusively licensed from MSK, which has been modified to enhance the degree and duration of the immune response against the WT1 protein. The modification is based on the fact that two of the four peptides in the peptide mixture comprising GPS are deliberately mutated in a single amino acid residue. These mutated peptides are recognized by the immune system as non-self entities and are therefore less likely to induce immune tolerance. After administration of these mutated peptides, the patients become immunized against the corresponding native versions of these peptides (which are expressed by the tumor cells), and thus, are able to cross-react against them, which concept is called the heteroclitic principle. The enhanced immunity and duration are largely independent of a patient’s HLA type. GPS also elicits both CD4 and CD8 immune responses. As described above, CD8 cells are extremely important, as their activation by GPS would lead to direct cancer cell killing, or cytotoxicity, and eventual establishment of immunologic memory against a WT1-expressing cancer. This occurs by two mechanisms, conversion of some of the activated CD8 cells to CD8 memory cells, and activation of CD4 cells and eventual creation of CD4 terminal effective memory cells.
Galena Biopharma is currently developing GPS for up to eight indications.
GPS monotherapy. GPS has completed Phase 2 clinical trials and has Phase 3 clinical trials planned (pending funding availability) for AML and MPM is also in various development phases as a potential treatment for MM and ovarian cancer. There is also an ongoing Phase 2 clinical trial of GPS for MM as monotherapy. The company also have plans to pursue additional clinical development programs for GPS as a monotherapy, including in CML and AML treated with hypomethylators.
GPS combination therapy. In October 2017, the company announced a clinical trial collaboration and supply agreement through a Merck subsidiary to conduct a combination clinical trial (using GPS along with the PD1 blocker pembrolizumab (Keytruda)) targeting up to five cancer types, namely colorectal cancer, small cell lung cancer, triple negative breast cancer, ovarian cancer and AML treated with hypomethylators. Galena Biopharma is preparing to start this clinical trial pending funding availability. Separately, a clinical trial in ovarian cancer of GPS in combination with nivolumab (Opdivo) is being conducted as an independently-sponsored trial by MSK. Finally, the company also have GPS delivery technology in preclinical development using licensed technology from Advaxis using a bacterial vector, Lm (which if successful, could lead to a second-generation product called WT1-Lm).
Potential Key Differentiators
GPS’ potential key differentiators as compared to other active immunization or vaccine-type approaches, as well as compared to immunotherapy approaches more generally, are as follows:
- heteroclitic peptides may offer increased immune response and less potential for tolerance;
- multivalent oligopeptide mixture potentially drives differentiated immunotherapeutic efficacy, targeting 25 key epitopes of WT1;
- potentially applicable to 20 or more cancer types worldwide and the vast majority of HLA types;
- CRem or minimal residual disease status (after initial tumor debulking with preceding standard therapy) is the preferred setting for GPS monotherapy;
- does not directly compete with current clinical standard of care therapies, but rather complements them in the maintenance setting;
- potential for combination approaches with other cancer immunotherapies, due to tolerable adverse event profile;
- anticipated cost-effective manufacturing; allogeneic, “off-the-shelf,” vialed subcutaneously administered drug that is not patient-specific; and
- positive Phase 2 clinical data on effectiveness (based on overall survival, or OS, in AML and progression-free survival, or PFS, in MM) with good tolerability and an innocuous safety profile.
Mechanism of Action
GPS has a mechanism of action that involves direct activation of the patient’s immune system specifically and solely against the WT1 protein. Typically, patients harboring WT1-positive malignancies have very few or no T-lymphocytes specifically reactive or responsive to, and therefore activated by, WT1. WT1 is a “self” antigen, against which the immune system is non-reactive, or said to be in a state of immune tolerance. Even if some patients have some innate T-cell responses naturally, these responses are weak and not adequate for any anti-cancer effect.
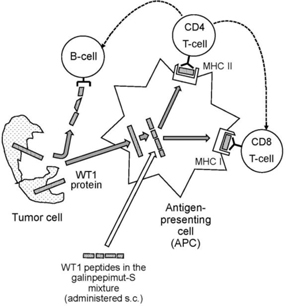
GPS is a WT1 peptide mixture. It cannot be administered to patients in a water-soluble form, and so it is given under the skin, or subcutaneously. If administered on its own, GPS would rapidly degrade and would not have the opportunity and the necessary time interval to activate the immune system. Therefore, GPS is mixed with Montanide, creating a dense emulsion. Additionally, prior to the administration of GPS, patients receive an adjuvant, GM-CSF to non-specifically stimulate and activate APCs in the vicinity of the subcutaneous injection of GPS.
After subcutaneous injection, the WT1 peptides within GPS disperse locally underneath the injection site and at local lymph nodes, and are ingested by APCs. Digested peptide fragments are then presented on the surface of APCs to CD8 and CD4 lymphocytes while simultaneously associated on the cell membrane with MHC/HLA molecules. This process activates the CD4 and CD8 cells and sensitizes them to the key 25 epitopes of WT1, thus initiating the process of short- and long-term T-cell-mediated immunity against WT1. CD8 cells then circulate around the lymphatic system and blood stream throughout the patient’s body targeting WT1-positive cancer cells. The stimulated CD8 cells transform into cytotoxic T-lymphocytes, or CTLs, which are able to attack and destroy specifically WT1-positive cancer cells. Each CTL typically destroys one WT1-positive cancer cell, but they have been shown to be able to kill up to 10 to 20 WT1-positive cancer cells. Further, CD4 cells are stimulated to produce WT1-specific, helper T-cells, which are able to in turn activate CTLs and B-cells. The B-cells “helped” by the helper T-cells produce antibodies to specific WT1 epitopes. The anti-cancer effect is considered to be a result of a combination of all of the above actions, as well as possible additional, less clear mechanisms involving other immune cell types (e.g., natural killer cells). The principles behind the above described mechanism of action of GPS are well established for the class of peptide-based active immunizing therapies of the vaccine type.
The following diagram illustrate GPS’ mechanism of action:
Targeted Indications
GPS Monotherapy for Acute Myeloid Leukemia
AML is an aggressive and highly lethal blood cancer characterized by the rapid growth of abnormal white blood cells that build up in the bone marrow and interfere with the production of normal blood cells. Its symptoms include fatigue, shortness of breath, bruising and bleeding, and increased risk of infection. The cause of AML is unknown, and the disease is typically fatal within weeks or months if untreated. AML most commonly affects adults, and its incidence increases with age. Current treatments include chemotherapy, and some patients may receive a hematopoietic, or blood-forming, stem cell transplant, or HSCT. The goal of upfront therapy for AML is to achieve a state of CRem. CRem is defined per consensus criteria by the European Leukemia Net, whereby the hematologic and clinical features of the disease are no longer detected. In principle, an allogeneic HSCT is an immunotherapy used clinically and specifically in AML, which works in four stages:
- achievement of CRem with standard upfront therapy followed by additional very high-dose chemotherapy that completely destroys any remnant of the patient’s blood forming cells, including any residual AML malignant cells;
- selection of a sufficiently genetically similar donor (usually one of the patient’s close relatives), called a histocompatible donor;
- removal of blood-forming cells from the bloodstream of that donor; and
- infusion of these donor cells into the patient for eventual engraftment onto the patient’s bone marrow and eventual creation of a completely re-instituted blood-forming system to sustain life and long-term leukemia-free status for the patient.
Barring the successful completion of an allogeneic HSCT in AML, no therapies have been proven to accord any meaningful long-term benefit after patients achieve a CRem status. Without allogeneic HSCT, once the disease relapses, second-line therapies can be given, but these have very limited positive clinical impact to date and their benefit is transitory; this means that eventually essentially all AML patients who do not undergo an allogeneic HSCT succumb to AML or complications associated with it.
The overall treatment landscape for AML has remained static for decades, as numerous (at the time, novel) targeted and antiproliferative agents failed to yield meaningful long-term clinical benefits, including increments in survival.
The AML indication was chosen for first-in-human clinical studies of GPS for the following reasons:
- AML presents a clinical setting in which CRem status can be achieved with standard upfront therapy;
- the almost universal expression of WT1 in leukemic blasts, which are AML’s malignant cells, as well as leukemic stem cells, or LSCs, cells that are or become extremely resistant to standard chemotherapy or targeted agent approaches and which can be realistically eradicated only with immunotherapy methods (including allogeneic HSCT). LSCs have been shown to be susceptible to targeting by cytotoxic T-cells (CD8 and CD4 cells) stimulated against leukemia-associated antigens and the company predicted this would be the case for GPS;
- the fact that WT1 has been associated with the actual development of leukemia;
- the positive correlation between the level of expression of WT1 and the prognosis in AML;
- the fact that the level of expression of WT1 can be followed over time in patients during and after therapy, including immunotherapy, as a method of monitoring for minimal residual disease, or MRD;
- early evidence from mouse models that vaccination with peptides against select WT1 antigenic epitopes leads to detection of immune response;
- early evidence that human immunocytes sensitized ex-vivo to peptides contained in GPS were able to recognize naturally presented WT1 peptides on the surface of several leukemia cell lines;
- early anecdotal (at the time) clinical data showing antileukemic activity of WT1 monovalent vaccines in the Japanese population (albeit restricted to HLA-A*2401 type), as well as a dendritic cell vaccine in the Netherlands (independent of HLA haplotype);
- the high degree of unmet medical need in AML and the absence of an effective maintenance therapy over the decades after initial upfront induction until and immediately after achievement of CRem status, particularly in patients older than 60 years of age;
- a predictive assumption of very low to negligible degree of clinical toxicity with a WT1-targeted immunotherapy such as GPS, due to the fact that WT1 in normal, non-cancerous, tissues is both expressed at extremely low levels and limited in number of organs and tissues, but also due to the fact that WT1 fragments, or peptide epitopes, in normal cells are presented to host APCs in a different manner than are WT1 fragments produced in cancer cells; of note, WT1 expression in normal tissues of adults is limited to the podocyte layer of the glomerulus (kidney), Sertoli cells (testis), granulosa cells (ovary), decidual cells (uterus), mesothelial cells (peritoneum, pleura), mammary duct and lobule (breast), and blood-forming (hematopoietic) progenitor cells (CD34+ cells in the bone marrow); and
- the advent of modern immunotherapeutics in cancer and the promise of an innovative, off-the-shelf immunotherapy for AML, a disease that was associated with dearth of deep and sustained responses to checkpoint inhibitors.
Clinical Data—AML
In an initial pilot clinical trial in AML, a total of nine adult patients of all ages with de novo AML were treated with upfront standard chemotherapy and were able to achieve their first complete remission, or CRem1. Administration of GPS resulted in a median OS that was at least 35 months from the time of GPS administration. In this study, specifically for patients who were 60 yrs and older (n=5), median OS was at least 33 months from the time of GPS administration or approximately 43 months from the time of initial AML diagnosis. The mean time of follow-up was 30 months from the time of diagnosis at the time of this analysis for all patients. Of the eight patients tested for immunologic response, seven, or 87.5%, demonstrated a WT1-specific immune response.
In a subsequent Phase 2 clinical trial in AML, a total of 22 adult patients of all ages with de novo AML were treated with upfront standard chemotherapy and were able to achieve CRem1. Most patients also received one to four cycles of “consolidation” chemotherapy per standard AML treatment guidelines. GPS was then administered within three months from the completion of the consolidation chemotherapy regimen in up to 12 total doses: Six initial doses (priming immunization) followed by six additional “booster” immunizations over a total period of up to 15 months to qualifying patients (i.e., patients who were clinically stable and did not show disease recurrence after the first six injections). This Phase 2 clinical trial met its primary endpoint of an actual OS rate of at least 34%, measured three years into the clinical trial (i.e., percentage of patients alive after three years of follow-up). An actual OS rate of 47.4% was demonstrated at 3 years post-GPS treatment, exceeding historical published data of OS of 20% to 25% by 2.4- to 1.9-fold (or 240% to 190%), respectively.
GPS administration was also shown to improve OS in comparison to historical data in patients in CRem1. Administration of GPS resulted in a median OS that was poised to exceed 67.6 months from the time of initial AML diagnosis in patients of all ages, which represents a substantial improvement compared to best standard therapy. Only five of the 22 patients underwent allogeneic HSCT and an ad hoc statistical analysis failed to show a significant effect of the transplant upon OS (either in median survival times or survival rates at specific landmark time-points). GPS was well tolerated in this patient population, whose median age was 64 years old. Moreover, GPS elicited WT1-specific immune responses in 88% of patients, including CD4 and CD8 T-cell responses. Further, the heteroclitic principle was confirmed, in that immune responses were seen against the native version of the two mutated WT1 peptides within the GPS mixture. The results showed a trend in improved clinical outcomes in patients who mounted an immune response with GPS compared to those patients who did not. Importantly, a preplanned subgroup analysis for the cohort of 13 patients within the clinical trial who were 60 years of age or older demonstrated a median OS of 35.3 months from time of initial diagnosis. This is also a remarkably prolonged value, considering that comparable historical populations have a median OS ranging from 9.5 to 15.8 months from initial diagnosis, which represents a 2.25 to 3.75-fold improvement in OS as compared to these historical cohorts of broadly comparable patients.
An additional Phase 2 clinical trial of GPS was performed at the H. Lee Moffitt Cancer Center & Research Institute, or Moffitt. This Phase 2 trial included ten AML patients who had received first-line therapy for their disease, who then experienced relapse and were subsequently treated with second-line chemotherapy and achieved a second complete remission, or CRem2. This group of patients had a more advanced disease in comparison to those treated in the other Phase 2 clinical trials discussed above, and typically demonstrated a historical OS of less than ~8 months, even with post-CRem2 allogeneic HSCT. In the Moffitt trial, the efficacy of GPS (measured as median OS from the time of administration of a maintenance therapy to immediately after achievement of CRem2) was compared with that of “watchful waiting” in a cohort of 15 contemporaneously treated (but not matched by randomization) broadly comparable patients treated by the same clinical team at Moffitt. GPS administration resulted in a median OS of 16.3 months (495 days) compared to 5.4 months (165 days) from the time of achievement of CRem2. This was a statistically significant difference (P=0.0175). Two of 14 AML patients demonstrated relapse-free survival of more than one year. Both such patients were in CRem2 at time of GPS administration, with duration of their remission exceeding duration of their CRem1, strongly suggesting a potential benefit based on immune response mechanisms. GPS was well-tolerated in this clinical trial.
Planned Phase 3 Clinical Trial—AML
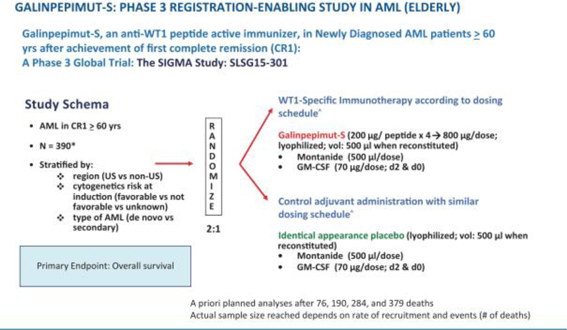
Galena Biopharma is planning a Phase 3 clinical trial for GPS in AML patients 60 years of age or older who have achieved CRem1 following upfront chemotherapy and up to two cycles of post-remission consolidation chemotherapy, but who will not undergo allogeneic HSCT. This clinical trial has been planned, a principal investigator and the majority of site investigators have been identified and its operational partners for the execution of the trial are in the process of being identified. After several meetings and correspondence exchanges, the FDA has indicated that the agency has no further comments on the clinical trial design, protocol or statistical analysis plan. In addition, well-qualified members of an independent data monitoring committee have agreed to join the independent data monitoring committee for this clinical trial upon its establishment.
The company currently plan to initiate this clinical trial, pending funding availability, in 2018.
The clinical trial is planned to include up to 180 centers in the United States, Canada, European Union, Eurasian Union, and other countries and an estimated total sample size of up to 390 patients. Randomization will be 2:1 (GPS:placebo) and on-trial treatment duration will be up to approximately 82 weeks (1.58 years). The primary endpoint of the clinical trial is OS, measured from the time of randomization (not initial diagnosis). No companion diagnostic will be used as AML universally expresses WT1. Randomization will be stratified by region (U.S. compared to non-U.S.), cytogenetic risk at diagnosis (favorable compared to not favorable compared to unknown), and type of AML (de novo compared to secondary). Patients will provide historical cytogenetic analysis results from initial diagnosis, before the start of their original chemotherapy treatment, to assess National Comprehensive Cancer Network genetic risk category. The clinical trial is currently powered to declare a positive result if GPS provides a 4-month OS advantage compared to placebo, namely increasing median OS from ~9 months in the control arm to ~13 months in the active, GPS-treated arm with an 1-sided α of 2.5%. Three interim analyses (IA1, IA2 and IA3) are planned in addition to a final analysis.
The following figure illustrates the AML Phase 3 GPS clinical trial schema described in the above paragraph.
GPS Monotherapy for Malignant Pleural Mesothelioma
MPM is an asbestos-related cancer that forms on the protective tissues that cover many of the internal organs. The most common area affected is the lining of the lungs and abdomen, though it can also form around the lining of the heart. Most cases are traced to job-related exposures to asbestos and it can take approximately 40 years between exposure and cancer formation. Symptoms may include shortness of breath, a swollen abdomen, chest wall pain, cough, feeling tired, and weight loss. MPM is generally resistant to radiation and chemotherapy, and long-term survival is rare, even in cases where aggressive upfront debulking multimodality therapy (i.e., extirpative surgery, chemotherapy and in some cases radiotherapy, often described as “trimodality therapy” when used to treat MPM) are used.
Assuming absence of distant, systemic metastatic disease, MPM patients can initially present with a very difficult-to-treat malignancy. The location, geometry, and origin of the tumor in the pleura (the external lining of the lungs and inner lining of the chest cage) present significant challenges for local and regional disease control. Extensive and complex surgery is initially considered and attempted to be planned. Patients without distant disease are broadly divided in two subgroups: (a) those who are in an inoperable status and (b) those who are operable. Patients in the former subgroup may be inoperable for two reasons: first, because they may be medically unfit for an extensive “definitive” surgery, most commonly due to co-morbidities (contemporaneously active diseases unrelated to their cancer) or, secondly, for technical reasons (location and/or bulk of tumor); the latter group of patients is defined as harboring “unresectable” disease. In general, approximately 35% to 40% of patients with a priori unresectable disease can be converted to technically resectable/marginally resectable, particularly if surgical expertise is high, after several cycles of upfront chemotherapy. This preoperative chemotherapy is termed “neoadjuvant” therapy. After the patient’s tumor becomes technically resectable, they receive extirpative surgery, often followed by more chemotherapy and sometimes radiotherapy. On the other hand, patients who are a priori operable proceed immediately with definitive surgery, resulting in either R0 or R1 resections, the degree between the two being assessed by surgical pathology review, with R0 corresponding to resection for “curative intent”, and R1 corresponding to microscopic residual tumor despite complete eradication by visual inspection at the time of surgery. After surgery, this subgroup of patients receives several cycles of chemotherapy and sometimes radiotherapy. This is postoperative chemotherapy termed “adjuvant” therapy.
In essence, all MPM patients who receive successful upfront trimodality therapy (schema A: Upfront neoadjuvant chemotherapy, followed by definitive surgery, followed possible further additional chemotherapy and schema B: upfront definitive surgery followed by adjuvant chemotherapy) become free of residual detectable, macroscopic malignant deposits. Like AML patients who achieve CRem after upfront chemotherapy (in the absence of allogeneic HSCT), virtually all MPM patients will eventually relapse. Recurrent disease is unfortunately minimally responsive to second-line chemotherapy in MPM and typically these patients succumb to their disease or related complications within a few weeks to months after the emergence of clinically evident recurrent MPM. To date, there is no effective maintenance type of therapy to delay or prevent MPM relapse after initially successful upfront trimodality therapy. Typical median OS, even when following a fairly aggressive regimen when surgery is feasible, is between 12 and 16 months following diagnosis. Nonetheless, highly select patients who both undergo R0/R1 extensive surgery and complete a full course (6 cycles) of indicated chemotherapy (specifically those receiving the combination of pemetrexed with cisplatin, either in the neoadjuvant or adjuvant setting) can survive up to 21.0 to 24.8 months following initial diagnosis. These patients are typically younger, in excellent functional status, without co-morbidities and possibly having tumor-related factors related to better prognosis, such as intrinsically higher sensitivity of MPM cancer cells to chemotherapy-induced destruction.
Like AML, MPM represents a “model” type of solid tumor for testing the effects of GPS in clinical studies for the following reasons:
- MPM presents a clinical setting whereby minimal residual disease status can be achieved with standard upfront therapy;
- the universal expression of WT1 in MPM malignant cells; in fact, WT1 expression is an established pathognomonic criterion for the actual diagnosis of MPM and its differentiation of other chest malignancies, for example, pulmonary adenocarcinoma;
- the positive correlation between the level of expression of WT1 and prognosis in MPM;
- preliminary evidence that WT1 expression could be involved in the MPM tumorigenesis and malignant growth promotion;
- early evidence that human APCs sensitized ex-vivo to peptides contained in the GPS mixture were able to recognize naturally presented WT1 peptides from MPM cell lysates;
- evidence that CD8 tumor-infiltrating lymphocytes predict favorable prognosis in MPM after resection (with the assumption that these CD8 cells are highly sensitized to tumor-associated antigens, including WT1);
- the high degree of unmet medical need in MPM and the absence of an effective maintenance therapy; indeed, despite extensive research efforts and recent promising, yet preliminary, results with checkpoint inhibitors in second or third line therapy of MPM patients, few options are available for the treatment of MPM in the maintenance setting after successful debulking with upfront trimodality therapy (with the vast majority being managed with “watchful waiting” until the disease’s inexorable relapse) and its prognosis remains very poor;
- a predictive assumption of very low to negligible degree of clinical toxicity with a WT1-targeted immunotherapy such as GPS, due to the fact that WT1 in normal, non-cancerous, tissues is both expressed at extremely low levels and limited in number of organs and tissues, but also due to the fact that WT1 fragments, or peptide epitopes, in normal cells are presented to host APCs in a different manner than are WT1 fragments produced in cancer cells; and
- an initial preliminary clinical efficacy “signal” from the Phase 2 clinical trial of GPS at MSK in patients with MPM.
Clinical Data—MPM
A randomized, double-blind, placebo-controlled Phase 2 clinical trial in MPM patients enrolled a total of 41 patients at MSK and M.D. Anderson Cancer Center. According to the Phase 2 MPM clinical trial data of GPS presented at the 2016 International Mesothelioma Interest Group and the 2016 Annual Meeting of the American Society of Clinical Oncology, as of May 2016, based on an initial analysis of 40 patients who were eligible at the time with a median follow-up of 16.3 months, a median OS of 24.8 months was recorded for GPS-treated MPM patients, compared to a median OS of 16.6 months for patients in the control arm, with a hazard ratio, or HR, of 0.51 in favor of GPS based on an initial analysis of 40 patients who were eligible at the time. Patients with an R0 tumor resection and subsequent treatment with GPS showed a significant survival benefit compared to those who received a placebo, with a median OS of 39.3 months compared to 24.8 months (HR: 0.415) in favor of GPS; this was a statistically significant difference (P<0.05). In a subsequent analysis of these endpoints for the entire cohort (n=41) in August 2016, with a median follow-up of 17.2 months, a median OS of 22.8 months was observed for GPS-treated MPM patients, compared to a median OS of 18.3 months for patients in the control arm, with an HR of 0.54 in favor of GPS. Furthermore, in the datasets from both of these analyses, GPS was shown to induce WT1-specific CD8 and CD4 T-cell activation. GPS administration in the 19 MPM patients in the active arm of the aforementioned study was commonly associated with mild (grade 1 and 2) and self-limited injection site reactions. Clinically significant severe adverse events did not occur.
Planned Phase 3 Clinical Trial—MPM
Galena Biopharma has planned a Phase 3 clinical trial in MPM, pending funding availability. The FDA has reviewed the clinical trial design in previous meetings and, following a formal end-of-phase 2 meeting, has indicated that the agency has no further comments on the clinical trial design, protocol or statistical analysis plan. Galena Biopharma is currently evaluating the best strategy to develop GPS in this indication.
The planned Phase 3 clinical trial may include up to 120 centers in the United States, European Union, and other countries and an estimated total sample size ranging from 120 to 500 patients. The sample size is variable due to the Bayesian statistical design of the clinical trial. Randomization will be 1:1 (GPS:placebo) and on-trial treatment duration will be up to 13 to 18 months. The primary endpoint of the clinical trial is OS, measured from the time of randomization (not initial diagnosis). No companion diagnostic will be used as MPM universally expresses WT1. Randomization will be stratified by region (U.S. compared to non-U.S.), timing of chemotherapy (neoadjuvant compared to adjuvant setting), and type of radiotherapy co-administered (intensity-modulated radiation therapy compared to other radiotherapy compared to none). The clinical trial will be adequately powered through a Bayesian adaptive approach to declare a positive result if certain a priori criteria are met, such as GPS providing an eight-month OS advantage compared to placebo, namely increasing median OS from approximately 16 months in the control arm to approximately 24 months in the active, GPS-treated arm with a 2-sided α of 5%; the exact values of the OS in the control and active arms (as well as the difference between the two) may differ from the above estimates so long as the “two look” group sequential, adaptive statistical design would be able to deliver at least 90% power with an one-sided α of 5% at the time of the definitive “positive signal” analysis. Two interim analyses (IA1, IA2) are planned (the second of the two, if positive for efficacy, would lead to main clinical trial early termination) in addition to a final analysis.
The following figure illustrates the MPM Phase 3 GPS clinical trial schema.

GPS Monotherapy for Multiple Myeloma
MM is a cancer formed by malignant plasma cells, and its cause is unknown. The overgrowth of plasma cells in the bone marrow crowds out normal blood-forming cells, causing low blood counts and anemia (a shortage of red blood cells). MM can also cause a shortage of platelets (cells responsible for normal blood clotting) and lead to increased bleeding and bruising, along with problems fighting infections due to low white cell counts and/or lower levels of infection-fighting antibodies. MM causes a host of organ problems and symptoms, including fatigue, bone pain, fractures, circulatory problems (in small vessels of the brain, eye retina, heart, bowel, etc.) and kidney failure.
Treatment for MM includes chemotherapy, glucocorticoids, drugs that modulate the immune system (immunomodulatory drugs, or IMiDs), radiation and autologous stem cell transplants, or ASCTs. Recently, several novel targeted agents, such as proteasome inhibitors and immunotherapeutics have been introduced in the treatment paradigms for MM. Most therapies in MM are applied in combination, sometimes with usage of three to four or even five agents administered concomitantly or sequentially. This has led to a progressive increase in the number of “lines” of therapy that MM patients receive, which currently can reach up to five to six or even higher. Of note, ASCT can be used more than once, called tandem ASCTs, to debulk the disease and offer prolonged secondary remissions. Finally, allogeneic HSCT is rarely used in MM, but still has its use in selected high-risk patients who are or become refractory to antimyeloma therapies.
The prognosis in MM is highly variable and depends on numerous risk factors, some related to the biology of the disease, others to the host (e.g., age and functional status). Consequently, median survival can vary from up to at least 15 years in non-high-risk patients who achieve CRes, as defined by the International Myeloma Working Group, or IMWG, criteria, to approximately three years (from time of initial treatment) in patients with MM who achieve less than partial response, or PR, after ASCT. There are patients with MM who fare even more poorly than described above, for example those in the immediately aforementioned group who also have high-risk cytogenetics at baseline who may survive on average less than three years. Similarly, patients who are ineligible for ASCT and are managed only with chemotherapy and long-term IMiD maintenance (with up to nine cycles of lenalidomide) who also achieve less than CRes and remain MRD-positive demonstrate a three-year OS rate of only about 55%; these landmark three-year OS rates decrease by approximately 40 to 50% in patients who also have high-risk cytogenetics at baseline. Despite significant therapeutic advances in the management of MM, the prognosis of patients with high risk cytogenetics at the time of diagnosis remains quite poor, even when they successfully complete an ASCT, particularly if such patients continue to have evidence of MRD.
MM represents an intriguing opportunity to study both the clinical and immunologic effects of GPS in a hematologic malignancy. Therapeutic targeting of WT1 through immune pathways has largely not been pursued by others to date, and this indication presents an opportunity to target a malignancy that remains “incurable” in a strict sense, even in the face of significant advances that have accorded significant survival and freedom-from-active-disease benefit in standard risk patients. MM was chosen as a target indication for GPS for the following reasons:
- a clinical setting whereby MRD status can be achieved with standard upfront therapy. In this indication, with induction therapy using modern combination regimens followed by melphalan conditioning for myeloablation and a successful autotransplant, MM patients can achieve either CRes or very good partial response per IMWG criteria. This subgroup of patients would be optimal candidates for GPS therapy, even if they remain MRD
 by flow cytometry or molecular markers;
by flow cytometry or molecular markers; - the detectable expression of WT1 in MM cells (malignant plasmacytes). In the past, MM was considered not to be a tumor type with strong expression of WT1. This was due to the use of immunohistochemical staining analysis with anti-WT1 antibodies that had suboptimal diagnostic sensitivity. It has been recently shown that while WT1 is expressed at lower levels in MM compared to other hematologic and solid tumors, this expression is almost universally seen and is highly relevant from an immunobiological perspective, as the immune system is able to reliably raise vigorous and sustained WT1-specific responses against malignant plasmacytes in the context of both MM and the rare, very aggressive variant of plasma-cell leukemia;
- preliminary evidence that WT1 expression could be involved in the MM tumorigenesis and promotion;
- early anecdotal (at the time) clinical data showing anti-myeloma activity of WT1 monovalent vaccines in Japanese patients (albeit restricted to HLA-A*2401 type);
- the high degree of unmet medical need in MM patients with high-risk cytogenetics who also remain MRD after frontline induction therapy and successful autotransplant, even when maintenance therapy is applied with either bortezomib or IMiDs (thalidomide); this has been shown in multiple studies, and to-date few options are available for addition of effective therapies in the maintenance setting to be added to agents such as lenalidomide (which is now standard of care in this setting); and
- a predictive assumption of very low to negligible degree of clinical toxicity with a WT1-targeted immunotherapy such as GPS due to the fact that WT1 in normal, non-cancerous tissues is both expressed at extremely low levels and limited in number of organs and tissues, but also due to the fact that WT1 fragments, or peptide epitopes, in normal cells are presented to host APCs in a different manner than are WT1 fragments produced in cancer cells.
Clinical Data—MM
Galena Biopharma has reported comprehensive Phase 2 data for GPS in 19 patients with MM, which indicate promising clinical activity among MM patients with high-risk cytogenetics at initial diagnosis who also remain at least MRD![]() after successful frontline therapy (induction regimen followed by ASCT). This subgroup of MM patients, when serially assessed per IMWG criteria, typically relapse/progress within 12 to 14 months after ASCT, even when they receive maintenance therapy with IMiDs such as thalidomide or proteasome inhibitors such as bortezomib. Of note, 18 of the 19 patients received lenalidomide maintenance starting after the first three GPS administrations following ASCT; the remaining single patient received bortezomib under the same schedule. All patients had evidence of at least MRD after ASCT, while 15 of the 19 also had high-risk cytogenetics at diagnosis. Combined, these characteristics typically result in low PFS rates that do not exceed 12 to 14 months following ASCT, even while on maintenance therapy with IMiDs or proteasome inhibitors, which are the current standards of care. As of June 2017, median PFS with GPS was 23.6 months, while median OS had not been reached. The company's results compare favorably with an unmatched cohort of broadly comparable MM patients with high-risk cytogenetics published by the Spanish PETHEMA group from the PETHEMA Network No. 2005–001110–41 trial. The company's GPS therapy demonstrated a 1.87-fold increase in median PFS, as well as a 1.34-fold increase in the PFS rate at 18 months compared to the aforementioned historical cohort, which included MM patients with high-risk cytogenetics and MRD
after successful frontline therapy (induction regimen followed by ASCT). This subgroup of MM patients, when serially assessed per IMWG criteria, typically relapse/progress within 12 to 14 months after ASCT, even when they receive maintenance therapy with IMiDs such as thalidomide or proteasome inhibitors such as bortezomib. Of note, 18 of the 19 patients received lenalidomide maintenance starting after the first three GPS administrations following ASCT; the remaining single patient received bortezomib under the same schedule. All patients had evidence of at least MRD after ASCT, while 15 of the 19 also had high-risk cytogenetics at diagnosis. Combined, these characteristics typically result in low PFS rates that do not exceed 12 to 14 months following ASCT, even while on maintenance therapy with IMiDs or proteasome inhibitors, which are the current standards of care. As of June 2017, median PFS with GPS was 23.6 months, while median OS had not been reached. The company's results compare favorably with an unmatched cohort of broadly comparable MM patients with high-risk cytogenetics published by the Spanish PETHEMA group from the PETHEMA Network No. 2005–001110–41 trial. The company's GPS therapy demonstrated a 1.87-fold increase in median PFS, as well as a 1.34-fold increase in the PFS rate at 18 months compared to the aforementioned historical cohort, which included MM patients with high-risk cytogenetics and MRD![]() post-ASCT and on continuous intensive maintenance with thalidomide +/- bortezomib. The company's Phase 2 clinical trial started in June 2014 and has enrolled a total of 20 patients of which 19 are currently evaluable. The safety profile was devoid of grade 3/4/5 treatment-related adverse events. All non-progression events were confirmed and ongoing as of the time of the latest presentation (median follow-up at 20 months for survivors). Immune response data showed that up to 91% of patients had successfully developed T-cell (CD8 or CD4) reactivity to any of the 4 peptides within the GPS mixture, while up to 64% of patients demonstrated immune response positivity (CD4/CD8) against more than 1 WT1 peptide (multivalent responses). Moreover, multifunctional cross-epitope T-cell reactivity was observed in 75% of patients to antigenic epitopes against which hosts were not specifically immunized, in a pattern akin to epitope spreading. Further,a distinctive link was shown between the evolution of immune responses and changes in clinical response status (achievement of CR/very good partial response clinical status per IMWG criteria) over time following treatment with GPS, with each patient being used as his or her own control for each longitudinal comparison. This association has not been previously described for a peptide vaccine in MM. In summary, the results offer mechanistic underpinnings for immune activation against WT1 in patients with aggressive, high-risk MM, and support the potential antimyeloma activity of GPS.
post-ASCT and on continuous intensive maintenance with thalidomide +/- bortezomib. The company's Phase 2 clinical trial started in June 2014 and has enrolled a total of 20 patients of which 19 are currently evaluable. The safety profile was devoid of grade 3/4/5 treatment-related adverse events. All non-progression events were confirmed and ongoing as of the time of the latest presentation (median follow-up at 20 months for survivors). Immune response data showed that up to 91% of patients had successfully developed T-cell (CD8 or CD4) reactivity to any of the 4 peptides within the GPS mixture, while up to 64% of patients demonstrated immune response positivity (CD4/CD8) against more than 1 WT1 peptide (multivalent responses). Moreover, multifunctional cross-epitope T-cell reactivity was observed in 75% of patients to antigenic epitopes against which hosts were not specifically immunized, in a pattern akin to epitope spreading. Further,a distinctive link was shown between the evolution of immune responses and changes in clinical response status (achievement of CR/very good partial response clinical status per IMWG criteria) over time following treatment with GPS, with each patient being used as his or her own control for each longitudinal comparison. This association has not been previously described for a peptide vaccine in MM. In summary, the results offer mechanistic underpinnings for immune activation against WT1 in patients with aggressive, high-risk MM, and support the potential antimyeloma activity of GPS.
GPS Combination Therapy with PD-1 blocker (nivolumab) for Ovarian Cancer
Epithelial cancer of the ovary, or ovarian cancer, is a relatively common gynecologic cancer that develops insidiously, and hence is associated with vague or no symptoms that would urge patients to seek medical attention. Not surprisingly, most women with ovarian cancer present with advanced (at least locally or regionally, and often systemically spread) disease. Ovarian cancer is managed with initial surgical resection followed by platinum-based chemotherapy. During the past decade, incremental advances in chemotherapy, and the introduction of targeted therapies (such as poly-ADP-ribose polymerase inhibitors and several others) and specially formulated compounds (such as liposomal anthracyclines) have resulted in improved survival and in more effective treatment of relapsed disease. In addition, a better understanding of genetic risk factors, along with aggressive screening, has permitted a tailored approach to preventive strategies, such as bilateral salpingo-oophorectomy in selected women along in specific patient populations genetically predisposed to this cancer (such as those harboring genetic alterations of the BRCA gene family). Although a complete clinical remission following initial chemotherapy can be anticipated for many patients, a review of “second-look” laparotomy, when it was often performed as a matter of routine care, indicates that less than 50% of patients are actually free of disease. Furthermore, nearly half of patients with a negative “second-look” procedure relapse and require additional treatment. Many patients will achieve a second complete clinical response with additional chemotherapy. However, almost all patients will relapse after a short remission interval of nine to 11 months. Effective strategies, such as introduction of novel immunotherapies, to prolong remission or to prevent relapse are required, as subsequent remissions are of progressively shorter duration until chemotherapy resistance broadly develops, leading to eventual disease-related demise.
Ovarian cancer represents an intriguing opportunity to study both the clinical and immunologic effects of GPS in another solid tumor. Additionally, therapeutic targeting of WT1 through immune pathways has largely not been pursued by others to date for this indication and ovarian cancer remains “incurable” once it advances and becomes disseminated, even in the face of significant advances in the field. Ovarian cancer was chosen as a target indication for the following reasons:
- ovarian cancer presents a clinical setting whereby MRD status can be achieved with standard upfront therapy both immediately after first line therapy, but also after effective debulking of the “first relapse.” The latter subgroup of patients (after successful second line treatment/first salvage, lacking demonstrable macroscopic residual disease) would be optimal candidates for GPS therapy, as no standard maintenance therapy exists for such patients and the subsequent relapse patterns and metrics are known and predictable;
- the high levels of expression of WT1 in ovarian cancer cells. In fact, WT1 expression is so frequent that pathologists routinely use immunohistochemical stains for WT1 (with a standardized convention for describing expression and determining as “positive” or “negative”) to help distinguish epithelial ovarian cancers from other tumors;
- preliminary evidence that WT1 expression may be linked to prognosis in ovarian cancer and that it may play an anti-apoptotic role in ovarian cancer cell lines;
- the high degree of unmet medical need in ovarian cancer patients after first (or subsequent) successful “salvage” debulking therapy and the absence of effective therapies for such patients; and
- a predictive assumption of very low to negligible degree of clinical toxicity with a WT1-targeted immunotherapy such as GPS due to the fact that WT1 in normal, non-cancerous tissues is both expressed at extremely low levels and limited in number of organs and tissues, but also due to the fact that WT1 fragments, or peptide epitopes, in normal cells are presented to host APCs in a different manner than are WT1 fragments produced in cancer cells.
Clinical Data—Ovarian Cancer
GPS is being studied in combination with nivolumab, a PD-1 immune checkpoint inhibitor, in an open-label, non-randomized Phase 1/2 clinical trial, which is independently sponsored by MSK. The aim of the study is to evaluate the safety and efficacy of this combination in patients with recurrent ovarian, fallopian tube or primary peritoneal cancer who are in second or greater clinical remission (after their successful first or subsequent “salvage” therapy). This Phase 1/2 clinical trial was planned to enroll at least ten patients with recurrent ovarian cancer who are in second or greater clinical remission at MSK. Patients enrolled in the clinical trial received the combination therapy during the clinical trial’s 14-week treatment period. Individuals who had not progressed by the end of this period also received a maintenance course of GPS. Initial immune response and clinical evolution data are due in the first half of 2018, as is information on the primary endpoint of this clinical trial, which is the safety of repeated GPS administrations, for a total of six doses, in combination with seven infusions of nivolumab. This clinical trial addresses the safety of GPS when co-administered with a checkpoint inhibitor, with the goal of possibly detecting an efficacy signal based on PFS and OS (versus historical data of monotherapy with nivolumab in this patient population), as well as documenting the pattern of WT1-specific immune responses post-GPS. Pending the successful progress of this clinical trial a larger, follow-on, randomized clinical trial may be planned.
GPS Combination Therapy with PD1 blocker (pembrolizumab) for Other Cancers
In addition, given the potential immunobiologic and pharmacodynamic synergy between GPS and a PD1 blocker, as well as the prevalent expression of WT1 in five select tumor types (colorectal cancer, triple-negative breast cancer, small cell lung cancer, ovarian cancer and AML), the company entered into a clinical trial collaboration and supply agreement through a Merck subsidiary for the conduct of a combination clinical trial of GPS with pembrolizumab (Keytruda).
The purpose of this five-arm “basket” trial is to determine if the administration of GPS in combination with pembrolizumab has the potential to demonstrate clinical activity in the presence of macroscopic disease, where monotherapy with either agent would have a more limited effect. The negative influence of tumor microenvironment factors on the immune response is predicted to be mitigated by PD1 inhibition (by pembrolizumab) thus allowing the patients’ own immune cells to invade and destroy cancerous growth deposits specifically sensitized against WT1 (by concomitantly-administered GPS).
NeuVax™ (nelipepimut-S)
NeuVax (nelipepimut-S) is a cancer immunotherapy targeting HER2 expressing cancers. NeuVax is the immunodominant nonapeptide derived from the extracellular domain of the HER2 protein, a well-established and validated target for therapeutic intervention in breast and gastric carcinomas. The NeuVax vaccine is combined with GM-CSF (Sargramostim) for injection in between the layers of the skin epidermis, ie., intradermal administration. Data has shown that an increased presence of circulating tumor cells, or CTCs, may predict reduced DFS and OS, suggesting a presence of isolated micrometastases, not detectable clinically, but, over time, can lead to recurrence of cancer, most often in distant sites. After binding to the specific HLA molecules on antigen presenting cells, the nelipepimut-S sequence stimulates specific cytotoxic T lymphocytes, or CTLs, causing significant clonal expansion. These activated CTLs recognize, neutralize and destroy, through cell lysis, HER2 expressing cancer cells, including occult cancer cells and micrometastatic foci. The nelipepimut immune response can also generate CTLs to other immunogenic peptides through inter- and intra-antigenic epitope spreading.
NeuVax for Breast Cancer
According to NCI, over 230,000 women in the United States are diagnosed with breast cancer annually. While improved diagnostics and targeted therapies have decreased breast cancer mortality in the United States, metastatic breast cancer remains incurable. Approximately 75% to 80% of breast cancer patients have tissue test positive for some increased amount of the HER2 receptor, which is associated with disease progression and decreased survival. Only approximately 20% to 30% of all breast cancer patients-those with HER2 immunohistochemistry, or IHC, 3+ disease, or IHC 2+ and fluorescence in situ hybridization, or FISH, amplified-have a HER2 directed, approved treatment option available after their initial standard of care. This leaves the majority of breast cancer patients with low-to-intermediate HER2 expression (IHC 1+, 2+) with tumors that are not HER2-amplified by FISH ineligible for targeted therapy with trastuzumab and without an effective targeted treatment option to prevent cancer recurrence.
The company currently have two investigator-sponsored trials ongoing with NeuVax in combination with trastuzumab (Herceptin; Genentech/Roche). The combination of trastuzumab and NeuVax has been shown pre-clinically and in a pilot study to be synergistic. The company's Phase 2b trial is a multi-center, randomized, single-blinded, placebo-controlled trial in 275 HER2 1+/2+ breast cancer patients with positive nodes and/or TNBC. The study combines NeuVax and trastuzumab (Herceptin) in the adjuvant setting aiming to prevent recurrence or death. Tumors in these women show low levels of expression of HER2, as measured by IHC, i.e., at a level of either 1+ or 2+ and, hence, these patients are not considered candidates for Herceptin. Patients who are hormone receptor-negative and HER2 1+/2+ by IHC are currently defined as TNBC patients. Eligible patients are randomized to receive NeuVax + GM-CSF + trastuzumab or trastuzumab + GM-CSF alone. The primary endpoint of the study is DFS. Genentech/Roche is providing the trastuzumab and partial funding for this trial. Data presented in October 2016 demonstrated that this novel combination of trastuzumab and NeuVax with HER2 low-expressing patients is well tolerated and the cardiac effects of trastuzumab are not impacted by the addition of NeuVax. In February 2017, the DSMB reported that there were no safety concerns with the trial and the trial is not futile. The recommendation from the DSMB was to continue the trial with one revision to the statistical analysis plan regarding the timing of the pre-specified interim analysis. Given the lengthy duration of enrollment for the trial, the DSMB determined that the pre-specified interim efficacy analysis be moved up from 12 months to 6 months after the last patient is enrolled. Enrollment was completed and the interim efficacy analysis occurred in late March 2018, as reported by it in a press release dated April 2, 2018.
The interim efficacy analysis, conducted by an independent DSMB of the efficacy and safety data for the study in an overall population of 275 patients as well as the two primary study target patient populations (node-positive and TNBC) after a median follow-up of 19 months, demonstrated a clinically meaningful difference in median DFS in favor of the active arm (NeuVax + Herceptin), a primary endpoint of the study, with hazard ratios of 0.67 and 0.61 in the intent to treat and modified intent to treat populations (i.e., those who received at least one dose of vaccine or control) as well as a 34.9% and 39.5% reduction in relative risk of recurrence in the active versus control arms in the intent to treat and modified intent to treat populations, respectively. A clinically meaningful and also statistically significant difference was found between the two arms in the cohort of patients (n= 98) with TNBC, with a hazard ratio of 0.26 and a p-value of 0.023 in favor of the NeuVax + Herceptin combination. Similarly, a clinically meaningful and statistically significant difference was found between the two arms in favor of the combination in the cohort of patients not receiving hormonal therapy (n = 110), with a hazard ratio of 0.24 and a p-value of 0.009. This pre-specified interim analysis also showed an adverse event profile with no notable differences between treatment arms. This analysis confirmed the 2016 data showing that the addition of NeuVax to Herceptin did not result in any additional cardiotoxicity compared to Herceptin alone. Based on these results, and the DSMB’s recommendation, the company plan to expeditiously seek regulatory guidance by the FDA for further development of the combination of NeuVax + Herceptin in TNBC, considering the statistically significant benefit of the combination therapy seen in this population with large unmet medical need.
The company's second combination investigator sponsored trial is a Phase 2 in HER2 3+ breast cancer patients who have completed neoadjuvant therapy with an approved regimen that includes trastuzumab and failed to achieve a pathological complete response, meaning they have microscopic evidence of residual disease and are therefore at an increased risk of disease recurrence. This multi-center, prospective, randomized, single-blinded Phase 2 clinical trial is enrolled with approximately 100 patients with a diagnosis of HER2 3+ breast cancer who are HLA A2+ or HLA A3+ and are determined to be at high-risk for recurrence. High-risk is defined as having received neoadjuvant therapy with an approved regimen that includes trastuzumab but not obtaining a pathological complete response at surgery, or those who undergo surgery as a first intervention and are found to be pathologically node-positive. These high-risk patients are known to have higher recurrence rates than other HER2 3+ breast cancer patients. Eligible patients will be randomized to receive NeuVax + GM-CSF + trastuzumab or trastuzumab + GM-CSF alone. The primary endpoint of the study is disease-free survival. Funding for this trial was awarded through the Congressionally Directed Medical Research Program, funded through the Department of Defense, via a breast cancer research program breakthrough award. In February 2017, the DSMB reported that there were no safety concerns with the trial and the trial is not futile. The pre-specified interim safety analysis was also completed on n=50 patients and demonstrated that the agent is well tolerated with no increased cardiotoxicity associated with giving NeuVax in combination with trastuzumab. The recommendation from the DSMB was to continue the HER2 3+ trial unmodified.
A Phase 3 PRESENT (Prevention of Recurrence in Early- Stage, Node- Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) study enrolled 758 HER2 1+/2+ patients who are node-positive and HLA A2 or A3 positive. On June 27, 2016, the independent data monitoring committee recommended that the Phase 3 PRESENT clinical trial be stopped for futility. The PRESENT trial was stopped, and the company initiated an investigation into the causes of the recommendation. The company's analysis of the data showed that there was a separation of the curves, albeit not statistically significant, with the control arm performing better than expected and the NeuVax arm performing consistent with its protocol assumptions for the control group. Because the study was deemed futile, the company closed the PRESENT trial.
NeuVax for Ductal Carcinoma In Situ of the Breast
DCIS is defined by the NCI as a noninvasive condition in which abnormal cells are found in the lining of a breast duct and have not spread outside the duct to other tissues in the breast. DCIS is the most common type of breast neoplasm with malignant potential. In some cases, DCIS may become invasive cancer and spread to other tissues, and at this time, there is no way to know which lesions could become invasive. Current treatment options for DCIS include breast-conserving surgery and radiation therapy with or without tamoxifen, breast-conserving surgery without radiation therapy, or total mastectomy with or without tamoxifen. According to the American Cancer Society, in the United States, there were over 60,000 diagnoses of DCIS in 2015. Galena Biopharma is supporting an independent investigator-sponsored, or IST, Phase 2 trial to evaluate women diagnosed with DCIS who are HLA-A2 positive, who express HER2 at IHC 1+, 2+, or 3+ levels, and who are pre or post menopausal. Patients will be randomized to one of two arms: NeuVax plus GM-CSF or GM-CSF alone. The clinical study name is VADIS: Phase 2 Trial of Nelipepimut-S Vaccine in Women with DCIS of the Breast. The trial is sponsored and operationalized by the NCI, studying NeuVax’s potential clinical effects in earlier stage disease. The trial has an immunological endpoint evaluating NeuVax peptide-specific cytotoxic T lymphocyte (CTL; CD8+ T-cell) response in vaccinated patients.
NeuVax for Gastric Cancer
According to the NCI, gastric (stomach) cancer is a disease in which malignant (cancer) cells form in the lining of the stomach. Almost all gastric cancers are adenocarcinomas (cancers that begin in cells that make and release mucus and other fluids). Other types of gastric cancer are gastrointestinal carcinoid tumors, gastrointestinal stromal tumors, and lymphomas. Infection with bacteria called Helicobacter pylori is thought to be the cause of gastric cancer and age, diet, and stomach disease can affect the risk of developing gastric cancer. Gastric cancer is often diagnosed at an advanced stage because there are no early signs or symptoms and is the second-most common cancer among males and third-most common among females in Asia and worldwide with over 63,000 new cases a year in India, where an initial clinical trial of NeuVax is planned. Overexpression of the HER2 receptor occurs in approximately 20% of gastric and gastro-esophageal junction adenocarcinomas, predominantly those of the intestinal type. Overall, without regard to the stage of cancer, only approximately 28% of patients with stomach cancer live at least five years following diagnosis and new adjuvant treatments are needed to prevent disease recurrence.
The company currently have an agreement with Dr. Reddy’s Laboratories Ltd., or Dr. Reddy’s, to conduct a Phase 2 independent investigator-sponsored study in gastric cancer in India. To date, Dr. Reddy’s has not initiated the Phase 2 study with NeuVax.
GALE-301 and GALE-302
GALE-301 and GALE-302 are cancer immunotherapies that target FBP receptor-alpha. FBP is a well-validated therapeutic target that is highly over-express in ovarian, endometrial and breast cancers, and is the source of immunogenic peptides that can stimulate CTLs to recognize and destroy FBP-expressing cancer cells. Current treatments after surgery for these diseases are principally with platinum-based chemotherapeutic agents. These patients suffer a high recurrence rate and most relapse with an extremely poor prognosis. GALE-301 and GALE-302 are immunogenic peptides that consist of a peptide derived from FBP combined with GM-CSF for the prevention of cancer recurrence in the adjuvant setting. GALE-301 is the E39 peptide, while GALE-302 is an attenuated version of this peptide, known as E39’. Two early stage clinical trials have been completed with its FBP peptides in ovarian, endometrial, and breast cancers. In June 2016, the FDA granted two Orphan Drug Product Designations for the treatment (including prevention of recurrence) of ovarian cancer: One for GALE-301 (E39) and one for GALE-302 (E39’).
GALE-301 and GALE-302 in Ovarian Cancer
According to the NCI’s Surveillance, Epidemiology, and End Results, or SEER Program, new cases of ovarian cancer occur at an annual rate of 11.9 per 100,000 women in the United States, with an estimated 22,280 new cases and 14,240 deaths in 2016. Only 46.2% of ovarian cancer patients are expected to survive five years after diagnosis. Approximately 1.3% of women will be diagnosed with ovarian cancer at some point during their lifetime (2011-2013 data). The prevalence data from 2013 showed an estimated 195,767 women living with ovarian cancer in the United States. Due to the lack of specific symptoms, the majority of ovarian cancer patients are diagnosed at later stages of the disease, with an estimated 80% of women presenting with advanced-stage (III or IV) disease. These patients have their tumors routinely surgically debulked to minimal residual disease, and then are treated with platinum- and/or taxane-based chemotherapy. While many patients respond to this treatment regimen and become clinically free-of-disease, the majority of these patients will relapse. Depending upon their level of residual disease, the risk for recurrence after completion of primary therapy is approximately 70%. Unfortunately, for these women, once the disease recurs, treatment options are limited and the disease is most likely incurable.
GALE-401 (anagrelide controlled release)
GALE-401 contains the active ingredient anagrelide, an FDA-approved product, for the treatment of patients with MPNs to lower abnormally elevated platelet levels. The currently available immediate release, or IR, version of anagrelide causes adverse events that are believed to be dose and plasma concentration dependent and may limit the use of the IR version of the drug. Therefore, reducing the maximum concentration, or C max, and increasing the half-life of the drug is hypothesized to reduce the side effects, while preserving the efficacy, potentially allowing a broader use of the drug.
GALE-401 in Essential Thrombocythemia
ET is a myeloproliferative blood disorder and is characterized by the overproduction of platelets in the bone marrow. Elevated platelets alter the normal process of blood coagulation and can lead to thromboembolic events. About a third of patients are asymptomatic at the time of diagnosis. However, many patients develop symptoms during the course of the disease that affect the quality of life.
Multiple Phase 1 studies in 98 healthy subjects have shown GALE-401 reduces the C max of anagrelide and increases the half-life following oral administration, appears to be well tolerated at the doses administered, and to be capable of reducing platelet levels effectively. The Phase 1 program provided the desired PK/PD (pharmacokinetic/pharmacodynamic) profile to enable the initiation of the Phase 2 proof-of-concept trial. The Phase 2, open label, single arm, proof-of concept trial enrolled 18 patients in the United States for the treatment of thrombocytosis, or elevated platelet counts, in patients with MPNs. Final safety and efficacy data from this Phase 2 trial were presented in December 2015 and demonstrated a prolonged clinical benefit with a potentially improved safety profile.
Galena Biopharma has analyzed its data and the treatment landscape for MPNs, with a current focus on ET. Subject to completion of the manufacturing of the new formulation and other internal work, GALE-401 would be poised to advance into a Phase 3 clinical trial in ET patients who are intolerant or resistant to hydroxyurea. This trial is designed to compare GALE-401 (drug arm) versus best available therapy to include a sizable population of patients treated with anagrelide IR.
** Strategic Collaborations and License Agreements**
Although the company currently have a number of collaborations with corporate partners for the development of its product candidates in various territories worldwide, the following development collaborations are those that are most significant to it from a financial statement perspective and where significant ongoing collaboration activity exists.
Exclusive License Agreement—Memorial Sloan Kettering Cancer Center
In September 2014, the company entered into a license agreement with MSK, under which the company were granted an exclusive license to develop and commercialize MSK’s WT1 peptide vaccine technology. The MSK original license agreement was first amended in October 2015, further amended in August 2016, amended and restated in May 2017 and again amended and restated in October 2017. In connection with the entry of the original license agreement and its amendments, MSK was issued or assigned an aggregate of 4,846 ordinary shares of Private SELLAS common stock for the year ended December 31, 2017. These common stock shares were converted into its common stock shares upon the Merger.
Under the terms of the current amended and restated MSK license agreement, the company agreed to pay minimum royalty payments in the amount of $0.1 million each year commencing in 2015 and research funding costs of $0.2 million in each year and for three years commencing in January 2016. The company also agreed to pay MSK a mid-six digit amount over a one year period in exchange for MSK’s agreement to further amend and restate the MSK license agreement in October 2017, which resulted in the grant of rights to additional intellectual property to it and extension/relaxing of certain deadlines. In addition, to the extent certain development and commercial milestones are achieved, the company also agreed to pay MSK up to $17.4 million in aggregate milestone payments for each licensed product, and for each additional patent licensed product, up to $2.8 million in additional milestone payments. The company also agreed to pay MSK a tiered royalty in the mid-single digits in the event of commercial sales of any licensed products. The company also agreed to raise $25.0 million in gross proceeds no later than December 31, 2018. In the event the company do not raise such amount by December 31, 2018, MSK may terminate the license agreement after complying with the notice and cure periods of the agreement, or MSK may elect to receive additional common stock shares in an amount equal to 1.5% of its then fully diluted share capital, which would stay the right to terminate for a period of time.
Unless terminated earlier in accordance with its terms, the MSK license agreement as amended and restated, will continue on a country-by-country and licensed product-by-licensed product basis, until the later, of: (a) expiration of the last valid claim embracing such licensed product; (b) expiration of any market exclusivity period granted by law with respect to such licensed product; or (c) ten (10) years from the first commercial sale in such country.
Merck & Co., Inc. Clinical Trial Collaboration and Supply Agreement
In September 2017, the company entered into a clinical trial collaboration and supply agreement through a Merck subsidiary, whereby the company agreed with the Merck subsidiary to collaborate on a research program to evaluate GPS as it is administered in combination with their PD1 blocker pembrolizumab (Keytruda) in a Phase 1/2 clinical trial enrolling patients in up to five cancer indications, including both hematologic malignancies and solid tumors.
The Phase 1/2 clinical trial will utilize a combination of GPS plus pembrolizumab (Keytruda) in patients with WT1+ relapsed or refractory tumors. Specifically, the study is expected to explore the following cancer indications: colorectal (arm enriched in but not exclusive to patients with microsatellite instability-low), ovarian, small cell lung, triple-negative breast, and AML. This study will assess the efficacy and safety of the combination, comparing overall response rates and immune response markers achieved with the combination compared to prespecified rates based on those seen with pemrolizumab alone in comparable patient populations. The trial is anticipated to begin in the third quarter of 2018 (pending funding availability).
Advaxis, Inc. Research and Development Collaboration Agreement
In February 2017, the company entered into a research and development collaboration agreement with Advaxis whereby the company agreed to collaborate on a research program to evaluate, through a PoP trial, a clinical candidate comprised of the combination of Advaxis’ proprietary Lm-based antigen delivery technology and GPS. Unless terminated earlier in accordance with its terms, the Advaxis agreement will expire upon the earlier of: (a) completion of the PoP trial or (b) a decision by the parties to cease further development of the clinical candidate.
The Advaxis agreement provides for cost-sharing between the parties, with Advaxis being responsible for the costs of performing the research activities and filing any investigational new drug, or IND, cost-sharing for preparation of the IND, and the company being responsible for the costs (exclusive of product costs) of conducting the PoP trial. The company also agreed to make certain non-refundable milestone payments to Advaxis having an aggregate amount of up to $108.0 million, upon meeting certain clinical, regulatory and commercial milestones. In addition, if net sales exceed certain targets, the company agreed to make non-refundable sales milestone payments up to $250.0 million and royalty payments based on specific royalty rates, with a maximum rate capped at a percentage rate in the low teens if net sales exceed $1.0 billion.
The University of Texas M. D. Anderson Cancer Center and The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. License Agreement
In September 2006, the company acquired rights and assumed obligations under a license agreement between Apthera and The University Texas M.D. Anderson Cancer Center, or MDACC, and The Henry M. Jackson Foundation, or HJF, which granted it exclusive worldwide rights to an United States patent covering the nelipepimut-S peptide and several United States and foreign patents and patent applications covering methods of using the peptide as a vaccine. Under the terms of this license, Galena Biopharma is required to pay an annual maintenance fee, clinical milestone payments and royalty payments based on sales of NeuVax, or other therapeutic products developed from the licensed technologies.
Biovascular, Inc. Exclusive License Agreement
In December 2013, the company acquired worldwide rights to anagrelide controlled release, or CR, formulation, GALE-401, through its acquisition of Mills, LLC, or Mills its wholly owned subsidiary, GALE-401 contains the active ingredient anagrelide, an FDA-approved product that has been in use since the late 1990s for the treatment of MPNs. Mills entered into an exclusive license agreement with BioVascular, Inc., or BioVascular. The license agreement granted it an exclusive license to develop and commercialize anagrelide CR formulation. Under the terms of the license agreement and its amendments, Mills agreed to pay BioVascular, a mid-to-low single digit royalty on net revenue from the sale of licensed products, as well as, future cash milestone payments based on the achievement of specified regulatory milestones. Galena Biopharma is responsible for patent prosecution and maintenance.
In September 2017, Mills and BioVascular entered into an amendment to its exclusive license agreement to modify the certain terms of the license agreement, including but not limited to, ![]() eliminating the 3% royalty rate on annual net sales of $50.0 million and the 4% royalty now applies to annual net sales of up to $100.0 million, (ii) making an advance payment of approximately $0.4 million for the milestone related to the initiation of the Phase 3 clinical trial payable in two tranches with the first payment of $0.2 million payable on or before October 31, 2017 and the second payment of approximately $0.2 million payable 30 days after the consummation of the Merger but no later than December 31, 2017, (iii) adding a payment for a sublicense by Mills to a third party of 25% of any cash received for upfront fees or milestone payments if the sublicense is executed prior to first patient enrolled in the Phase 3 clinical trial and 17.5% of any cash received for upfront fees or milestone payments if the sublicense is executed after the first patient is enrolled in the Phase 3 clinical trial, and (iv) if the first patient is not enrolled in the Phase 3 clinical trial by December 31, 2018, BioVascular shall have the right to terminate the license agreement and the advance payment shall not be repaid to Mills. Under the terms of a September 2017 consent between Comerica Bank, BioVascular and Mills, Comerica Bank shall receive $0.1 million of the approximately $0.4 million advance payment from Mills.
eliminating the 3% royalty rate on annual net sales of $50.0 million and the 4% royalty now applies to annual net sales of up to $100.0 million, (ii) making an advance payment of approximately $0.4 million for the milestone related to the initiation of the Phase 3 clinical trial payable in two tranches with the first payment of $0.2 million payable on or before October 31, 2017 and the second payment of approximately $0.2 million payable 30 days after the consummation of the Merger but no later than December 31, 2017, (iii) adding a payment for a sublicense by Mills to a third party of 25% of any cash received for upfront fees or milestone payments if the sublicense is executed prior to first patient enrolled in the Phase 3 clinical trial and 17.5% of any cash received for upfront fees or milestone payments if the sublicense is executed after the first patient is enrolled in the Phase 3 clinical trial, and (iv) if the first patient is not enrolled in the Phase 3 clinical trial by December 31, 2018, BioVascular shall have the right to terminate the license agreement and the advance payment shall not be repaid to Mills. Under the terms of a September 2017 consent between Comerica Bank, BioVascular and Mills, Comerica Bank shall receive $0.1 million of the approximately $0.4 million advance payment from Mills.
Manufacturing
The company do not own or operate manufacturing facilities for the production of its product candidates nor do Galena Biopharma has plans to develop its own manufacturing operations in the foreseeable future. The company currently depend on third-party contract manufacturers for all of its required raw materials, active pharmaceutical ingredients, and finished product candidate for its clinical trials. The company do not have any current contractual arrangements for the manufacture of commercial supplies of any product candidates. The company currently employ internal resources and third-party consultants to manage its manufacturing contractors.




